


Pathophysiology of IPF

The cause of the condition is unknown, making it challenging to target with pharmaceutical drugs. It is hypothesized that recurrent microinjuries to the alveolar epithelium, perhaps through prolonged exposure to environmental pollutants or to viruses, leads to the release of pro-fibrotic cytokines, including TGF-β1, TNF-⍺, IL-6, and IL-8. This, in turn, activates underlying fibroblasts, causing them to adopt a myofibroblast phenotype characterized by excessive extracellular matrix (ECM) deposition. There is an urgent need for new drug targets and treatments, which includes new tools to better predict drug efficacy during preclinical drug discovery. REPROCELL is one of a handful of research groups and companies working to develop new models and preclinical test systems, which can accelerate the development of new treatments for IPF.
Key Application: Testing Drug Reponses in Profibrotic Stimulation Protocols
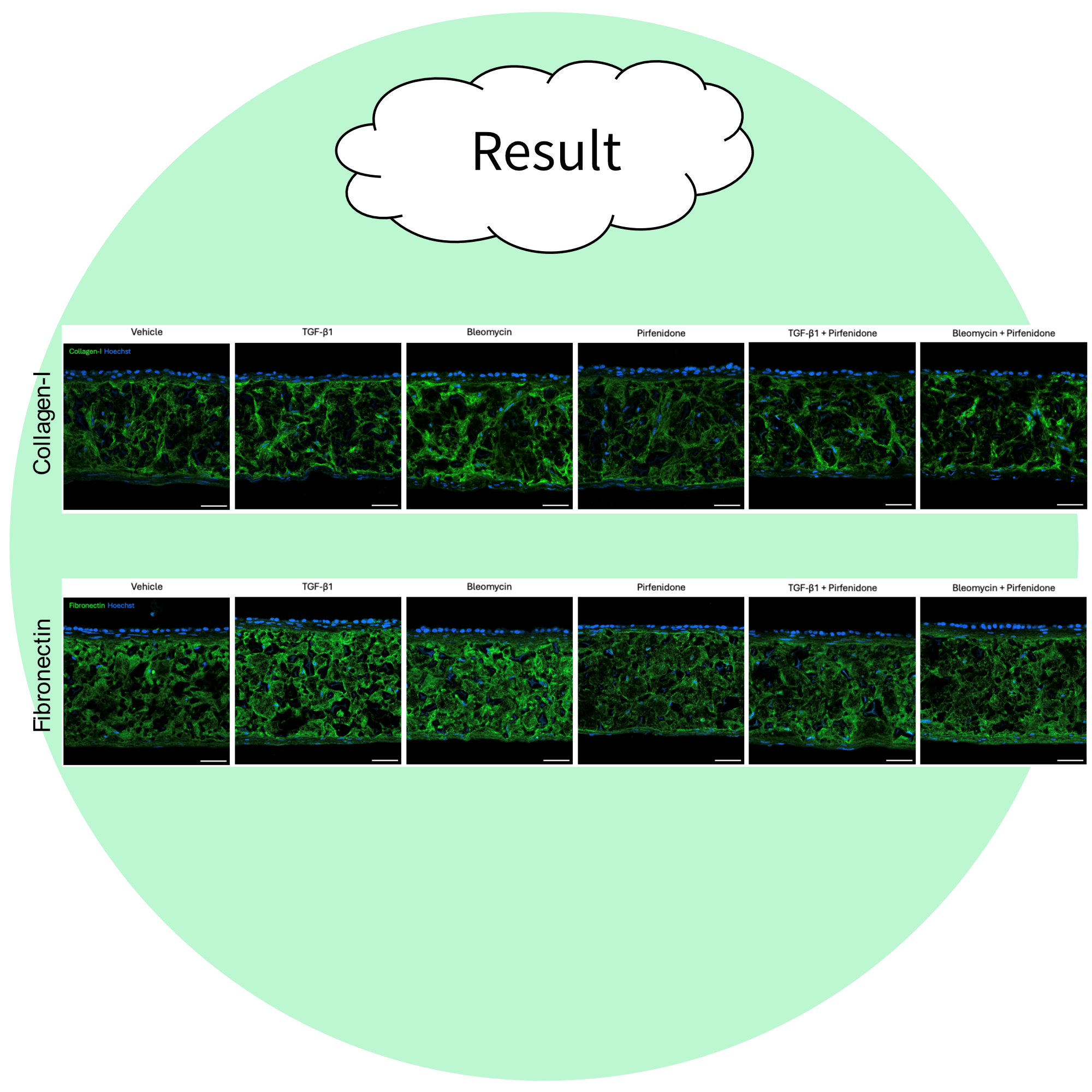
Total collagen and fibronectin is increased in the IPF lung models compared to the healthy controls (see below). In addition the effects of profibrotic stimuli such as TGF- β1 or bleomycin can be assessed in the model; both TGF-β1 and bleomycin are pro-fibrotic stimulants that are commonly used in models of IPF to initiate a fibrotic phenotype.

In our Alvetex model of IPF, TGF- β1 and bleomycin both lead to an increase in total collagen content in both healthy and diseased models, and alterations in the expression of matrix metalloproteinases (MMP) and tissue inhibitor of metalloproteinases (TIMP). Changes in the expression of MMP 1, MMP 3, and TIMP 2 in response to TGF-β1 or bleomycin in healthy and diseased lung models are shown below.

The effects of your test compound can be evaluated in the presence and/or absence of established profibrotic stimulators such as TGF-β1 or bleomycin (shown above) and can provide comparisons to the efficacy of standard of care drugs such as pirfenidone (below).
