Parkinson's Disease (PD) is a neurodegenerative condition that causes progressive degeneration of dopaminergic neurons in the central nervous system (CNS). There is currently no cure for PD, and standard-of-care treatments are limited in their efficacy; meaning new therapeutics are urgently required. However, conventional models used in drug development often lack clinical translatability.
In this case study, our scientists established a clinically-relevant in vitro model of Parkinson's disease using induced pluripotent stem cells (iPSCs). Read on to find out how they successfully developed this translational assay for drug screening – from patient screening to tissue collection, primary culture derivation, iPSC generation, and even using a medium as a co-culture alternative.
Preclinical models for Parkinson’s Disease
At REPROCELL, we strongly believe that human tissue testing is the most relevant way to explore drug behaviour prior to clinical trial. Fresh CNS tissues are an excellent alternative to conventional models but are notoriously difficult to obtain as they cannot be procured from living donors.
On the other hand, iPSCs can be donated by living patients which enables follow-up studies and improves tissue access. Because iPSCs can be transformed into a range of different neuronal cell types, autologous co-culture models can be created to improve the clinical relevance of these models even further.
How we established an in vitro Parkinson's disease model
Due to their unrivalled translatability, we wanted to develop an iPSC-derived in vitro model of PD for drug discovery. In the following section, we will walk you through each step of model establishment; from donor recruitment, iPSC generation, and the quality control checks performed. You can purchase the iPSCs we used to establish this model from REPROCELL.
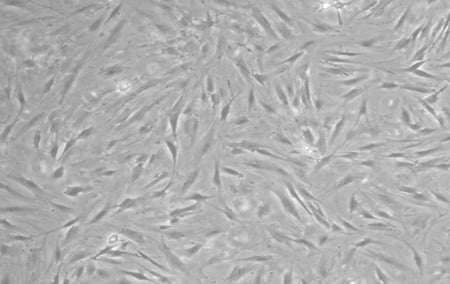
Step 1: Obtaining Primary Tissues
The reprogramming of iPSCs requires tissue from a consenting adult patient - to ensure that our model was as relevant as possible, we wanted to obtain primary tissues from a real PD donor. After locating a patient with a sporadic form of PD, a skin punch biopsy was collected, transported to our laboratory, and then used to produce a primary fibroblast culture. Below, we have included a phase contrast image of the fibroblasts derived from this PD donor.
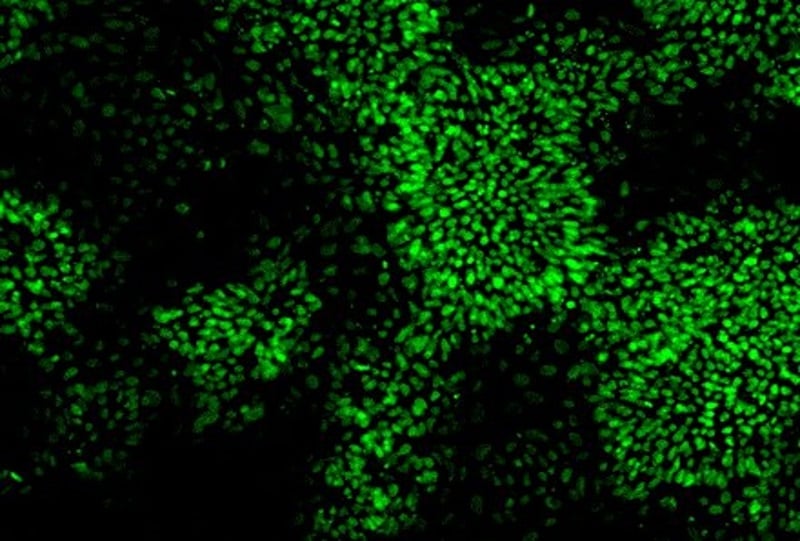
Step 2: Reprogramming primary tissue into stem cells
Following isolation of the fibroblasts, we reprogrammed them using StemRNA 3rd Gen Reprogramming Technology. This methodology was chosen as it negates the need to screen iPSC clones, and is 50 times more efficient than other non-integrative reprogramming kits*. The kit can be purchased here with a full reprogramming protocol available here.
Emergence of iPSC colonies was observed just seven days after reprogramming. Subsequent quality checks confirmed the potential of these cells to differentiate into any of the three germ layers, and stronger expression of stem cell markers than the EPCs and iPSC used in a control panel purchased from ThermoFisher1.
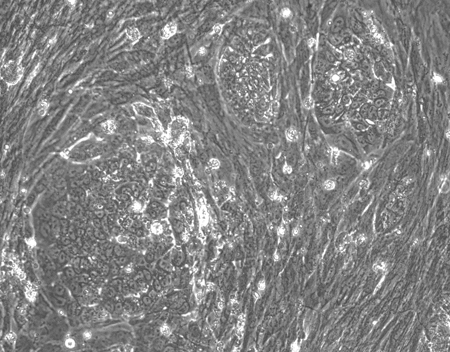 |
| Figure 2: Using StemRNA 3rd Gen Reprogramming Technology, we saw iPSC colonies emerging a week after reprogramming |
Step 3: Differentiating iPSCs into Neurons
After verifying the pluripotency, karyotype, and differentiation potential of our iPSCs, we proceeded to differentiate these cells in dopaminergic neurons using a protocol adapted from Kricks et al (2011)2. This methodology gives rise to dopaminergic neuronal progenitor cells around day 21 and fully mature neurons before day 60.
At day 65, we had produced a homogeneous population of dopaminergic neurons that expressed the neuronal marker Tuj1, and dopaminergic markers TH/PITX3 with very few glial cells present. Our dopaminergic neurons matured more quickly when cultured in MQ medium compared with astrocyte co-culture or basic differentiation media.
| A | %3B%20DAPI%20(blue).jpg?width=198&name=1%20Tuj1%20(red)%3B%20DAPI%20(blue).jpg) |
B | %3B%20DAPI%20(blue).jpg?width=200&name=2%20TH%20(green)%3B%20DAPI%20(blue).jpg) |
C | %3B%20Tuj1%20(red).jpg?width=200&name=3%20PITX3%20(green)%3B%20Tuj1%20(red).jpg) |
| Figure 3: A, Tuj1 (red), DAPI (blue); B, TH (green), DAPI (blue); C, PITX3 (green), Tuj1 (red) | |||||
Quality control procedures and regulatory requirements
Quality control for primary tissues
- Immunocytochemistry (ICC) to confirm the presence of fibroblasts in the primary culture.
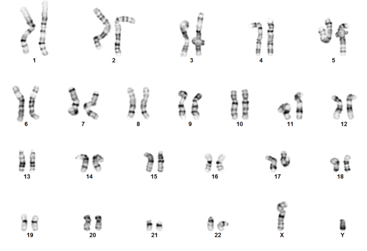
- G-banding to confirm normal karyotyping.
- Genetic profiling via STR analysis.
- Viral pathogen testing, sterility testing, and mycoplasma testing.
%3B%20DAPI%20(blue).jpg?width=344&name=Vimentin%20(green)%3B%20DAPI%20(blue).jpg)
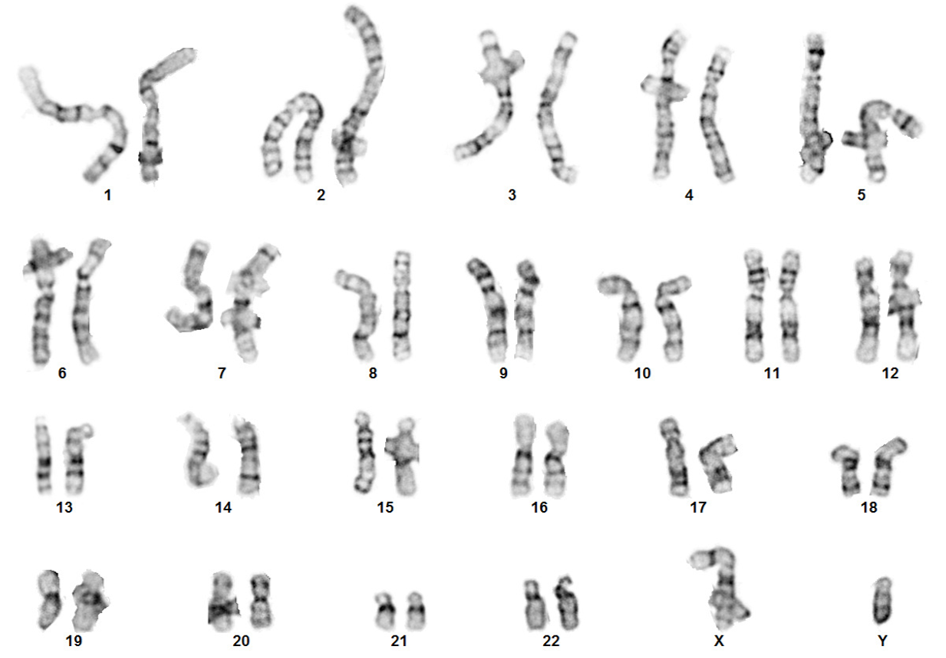
Regulatory considerations for primary tissues
- Confirm that patients/donors have been properly consented.
- Ensure that the donor clinical data is anonymized.
- Check that the appropriate MTA is in place if primary tissues are obtained externally
Quality control for reprogramming
- ICC of stem cell markers to verify pluripotency e.g. OCT4.
- G-banding to confirm normal karyotype.
- TaqMan hPSC Scorecard Assay1 to demonstrate pluripotency and trilineage differentiation potential.
- Genetic profiling to check that STR analysis is consistent with the parental fibroblast line.
%3B%20SSEA-4%20(red).jpg?width=390&name=Oct4%20(green)%3B%20SSEA-4%20(red).jpg)
Quality control for neuron differentiation
- ICC of dopaminergic and neuronal markers e.g. Tuj1, TH, PITX3.
- MEA analysis to verify neuronal activity of mature cell networks.
- RNA seq analysis to check maturity of dopaminergic neurons.
%3B%20GFAP%20(red)%3B%20DAPI%20(blue).jpg?width=500&name=Tuj1%20(green)%3B%20GFAP%20(red)%3B%20DAPI%20(blue).jpg)

A deeper look at culture optimization and analysis
Once we had successfully derived dopaminergic neurons from our iPSC culture, we moved onto optimizing the culture conditions for neuronal maturation. It was important to show that the neurons had matured successfully, but also that they displayed functionality. We used a range of analytical processes to ensure out culture conditions were optimal, including RNA seq and Micro-electrode array (MEA) analysis. The three culture conditions we explored for our neurons included:
- Basic dopaminergic medium
- MQ medium (RCDN102)
- Co-culture with iPSC-derived astrocytes (802-3G)
Co-culture of neurons with astrocytes
To produce the co-culture system, we had to differentiate a second control line into astrocytes. These cells were matured until day 95 where their astrocytic identity was confirmed with GFAP and CD34 staining. A physical co-culture would not be possible for RNA seq analysis (a pure dopaminergic neuron population was required) so we cultured the astrocytes in a trans-well above the dopaminergic neurons to avoid physical contact between the cells.
RNA seq analysis results
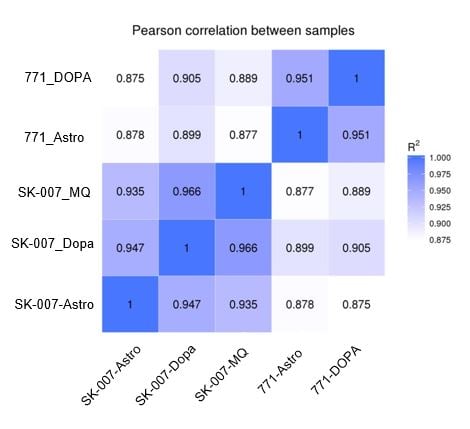
Results from the RNA seq analysis suggested that the genetic background of the cell line had a bigger impact on gene expression than the culture conditions. Less than 4% of genes were divergent between cells matured using different protocols.
 |
 |
|
Figure 7A: Pearson Diagram. The genetic background (control vs PD) is a bigger factor than the method of maturation (control medium vs coculture). |
Figure 7B: Venn diagram. The number of genes in common between culture conditions is extensive (less than 4% are divergent). |
Micro-electrode array analysis results
In preparation for MEA analysis, we plated our freshly differentiated neurons onto wells that each contained 16 electrodes. A true astrocyte/neuronal co-culture could be used for this experiment, meaning no transwells were required.
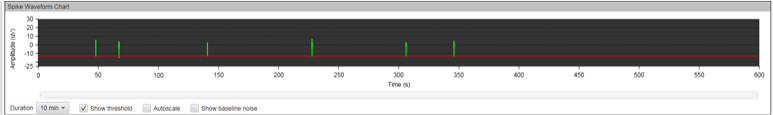
MEA recording was performed every seven days after the initial plating for a total of 12 weeks. During the first few weeks, no spontaneous neuronal firing was recorded in any of the culture conditions. After week three, we noticed an increased electrical activity in the wells containing MQ Medium and those with the co-cultured cells, but not all electrodes were firing.
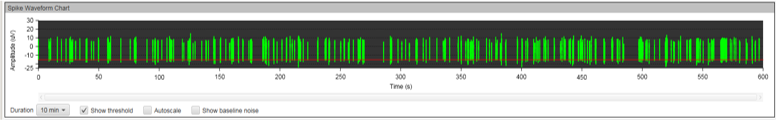
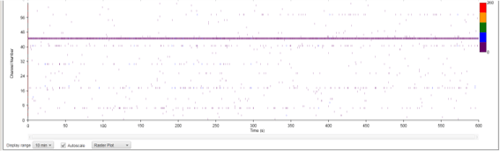
The results at week eleven were striking. While the co-cultured neurons displayed strong electrical activity, no synchronicities were present. Cells cultured in MQ medium displayed strong electrical activity AND the spikes were synchronous in the majority of electrodes. This is a phenomenon called bursting, which is characteristics of functional neuronal networks.
 |
||
 |
||
 |
||
|
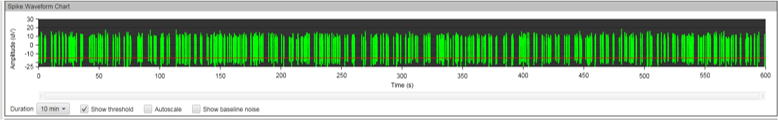
Figure 8A: MEA data at week three (raw data). Each green line represents a spike of activity over time, and each graph represents the recording of one electrode over time. Top, Dopaminergic medium; Middle, MQ Medium; Bottom, co-culture. |
||
 |
||
|
|
||
 |
||
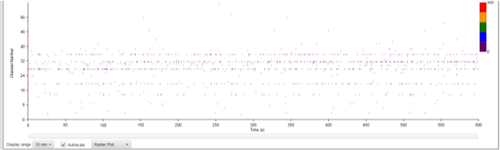
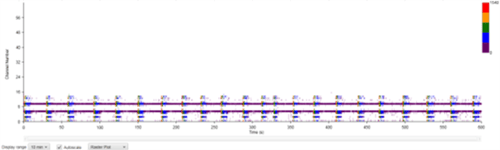
| Figure 8B: MEA data at week 11 (Rasta plots). Each group of 16 in the y axis represents the recording of one electrode, and each group on 16 in the y axis represents the recording of one well over time (x axis). Top, Dopaminergic medium; Middle, MQ Medium; Bottom, co-culture |
Neural Differentiation Services
Are you lacking the time or expertise to establish your in vitro PD model in-house? At REPROCELL, we offer neuronal differentiation projects that are fully customized to your unique research needs. Decades of experience in iPSC research has allowed us to develop robust differentiation protocols for dopaminergic, motor, and sensory neurons. We are also able to offer astrocyte differentiation for clients interested in advanced co-culture systems. We can help you at any stage of your disease model journey. Inquire today, and discover how we can help make your research goals a reality.
*StemRNA™ 3rd generation is at least 50 times more efficient (2.00 - 4.00%), compared with Episomal reprogramming methodology Epi5 (0.03 - 0.04%).
Editors note: This blog post was origionally published in March 2021 and has since been edited for accuracy and clarity.
Further reading


About the author
Zara Puckrin BSc, digital marketing manager, REPROCELL Europe
Zara is a GCU graduate who loves minimalism, marketing, and molecular biology. You can contact her on LinkedIn.
Subjects we write about
- 3D Cell Culture
- Cell Culture
- Central Lab Services
- Clinical Capabilities
- Disease Modeling
- Drug Discovery
- Gene Editing
- Genomic Services
- GMP
- Human Tissue Samples
- Human Tissue Testing
- IBD
- Life Sciences
- Master Cell Banks
- Neurons
- Oligonucleotide Synthesis
- Pharmacology-AI
- Precision Medicine
- Product Catalog
- Regenerative Medicine
- Respiratory Disease
- Safety Pharmacology
- Skin Disease
- Stem Cells
.jpg?width=400&height=225&name=New%20Approach%20Methodologies%20(NAMs).jpg)


.jpg?width=400&height=225&name=Untitled%20design%20(10).jpg)

