
The discovery of both CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) and associated proteins, the Cas nucleases, has revolutionized the field of gene-editing. Characterization of the role of the CRISPR-Cas system in pathogen detection, the immune response, and DNA repair mechanisms, has enabled this naturally occurring biological system to be harnessed and developed as a valuable research tool.
CRISPR-Cas technology has unlocked new and improved opportunities in biological and genetic research, food production, and increasingly, in the development of gene therapy and cell-based therapeutics. However, as with all new developments, the full potential of the technology can only be reached if it is scientifically valuable, commercially transferable, cost-effective, and user-friendly. Use of conventional CRISPR-Cas technology is limited for some gene-editing applications including simultaneous multiple gene knock-out, knock-in of longer length DNA fragments and single nucleotide modification.
Here, we highlight CRISPR-SNIPER technology, a timesaving CRISPR-Cas system with built-in optimization capacity and improved gene-editing efficiency that overcomes the limitations of conventional CRISPR-Cas and broadens application of the gene-editing technology. Delivered by REPROCELL, CRISPR-SNIPER takes gene-editing to new and improved levels of success.
Fundamentals of the CRISPR-Cas system
Alternative gene-editing technologies, such as TALENs (transcription activator-like effector nucleases) and zinc finger nucleases (ZFNs), paved the way for precise genome modification techniques. However, they have several limitations in use.
Primarily, TALEN-based systems differ from the CRISPR-Cas system in that they are protein-based DNA editing techniques whereas the CRISPR-Cas system is a small RNA-mediated sequence-specific cleavage technique. The ability of the CRISPR-Cas system to target a gene of interest using a specific guide RNA (sgRNA) means that the system is highly programmable and nucleotide specific – one of the key elements to its success as a research tool.
Generating sgRNA is a relatively straightforward laboratory process enabling the adaptation of CRISPR-Cas methods to many different applications. The technology was initially developed as a method to knock out target genes in functional studies, however, recent technical advances have extended this remit to selectively activate or repress target genes, purify specific regions of DNA, image DNA in live cells, and precisely edit DNA and RNA sequences.
The type II CRISPR-Cas9 system, the most commonly used system, has more target sites than ZFNs and TALENs, and numerous Cas9 variants which enhance its ability to perform systematic analyses of gene function and allow specific genome targeting in many different organisms, including mammalian cells.
The principle of the CRISPR system is simple: a non-coding RNA molecule guides the Cas9 nuclease to a gene sequence inducing a site-specific cleavage (Figure 1).
DNA damage is then repaired by cellular DNA repair pathways – non-homologous end joining repair (NHEJ) or homology-directed repair (HDR). The NHEJ pathway is efficient but error-prone, introducing insertions and deletions (INDELs) that may affect gene function. INDELS can then be characterized to identify and select the mutations of interest.
By contrast, the HDR pathway is less efficient but with high-fidelity making it useful to generate specific nucleotide changes ranging from a single nucleotide change to large insertions. The NHEJ pathway is naturally the preferred option for most cell types, and additional components are needed to drive the repair mechanism towards the more specific HDR pathway.
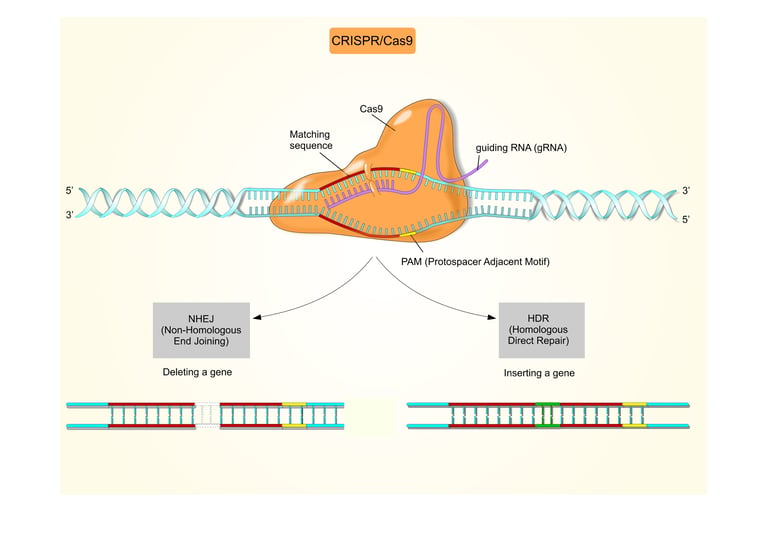 Figure 1.The CRISPR-Cas9 Complex. A short synthetic RNA containing a sequence required to bind Cas9 nuclease and a user defined sequence specific for the target guides the RNA/nuclease complex to the gene of interest. The target sequence must be unique within the genome and located near a protospacer adjacent motif (PAM) – a binding signal for Cas proteins. Binding of the sgRNA to Cas9 activates the nuclease activity and allows DNA cleavage to occur.
Figure 1.The CRISPR-Cas9 Complex. A short synthetic RNA containing a sequence required to bind Cas9 nuclease and a user defined sequence specific for the target guides the RNA/nuclease complex to the gene of interest. The target sequence must be unique within the genome and located near a protospacer adjacent motif (PAM) – a binding signal for Cas proteins. Binding of the sgRNA to Cas9 activates the nuclease activity and allows DNA cleavage to occur.
The basic CRISPR-Cas experimental workflow is a 3-stage process
- Design: Choose the best and/or specific components for a particular cell type e.g. sgRNA and Cas nuclease
- Delivery: Choose the best format (plasmid DNA, RNA, ribonucleoprotein) and delivery method (electroporation, lipofection, virus) to allow the editing process to take place within a cell
- Analysis: Characterize the edits using PCR and Sanger or Next Generation Sequencing methods to determine gene editing efficiency and select mutations of interest
The complete workflow can take several months, and each stage has its own set of challenges. Cas9 nuclease activity, target site selection and sgRNA design, delivery methods, off-target effects, and the incidence of HDR are all factors potentially affecting the efficiency and specificity of the system. Developments in informatics and machine learning help abrogate factors such as target selection and sgRNA design, and molecular modification of the Cas nucleases can increase activity.
However, even with these optimizations and improvements, the process can still take many months to optimize and complete, and the gene-editing efficiency plus the number of positively edited clones may still be inadequate. A key problem with conventional CRISPR-Cas is that experimental failure is only recognized after many weeks or even months of effort have been expended. Reducing timelines and improving editing efficiency are necessary if the technology to progress and develop.
Advancing CRISPR-Cas technology with CRISPR-SNIPER
CRISPR-SNIPER technology recently introduced by REPROCELL in collaboration with GenAhead Bio, takes the CRISPR-Cas system to the next level - streamlining timelines, reducing overall costs, and increasing the efficiency and potential application of the system. The technology addresses key areas of the 3-stage process – the analytical stage and the optimization steps required for the design and delivery stages.
The analytical stage of traditional CRISPR-Cas technology is generally the most onerous, labor-intensive, and costly stage, and often ends in failure. Following delivery and transfection of the system components to allow gene-editing to occur, a limiting dilution of the cells is performed, and individual clones then picked for analysis. Depending on the efficiency of the system, this process may be repeated several times, and on average, hundreds of clones selected and checked (Figure 2).
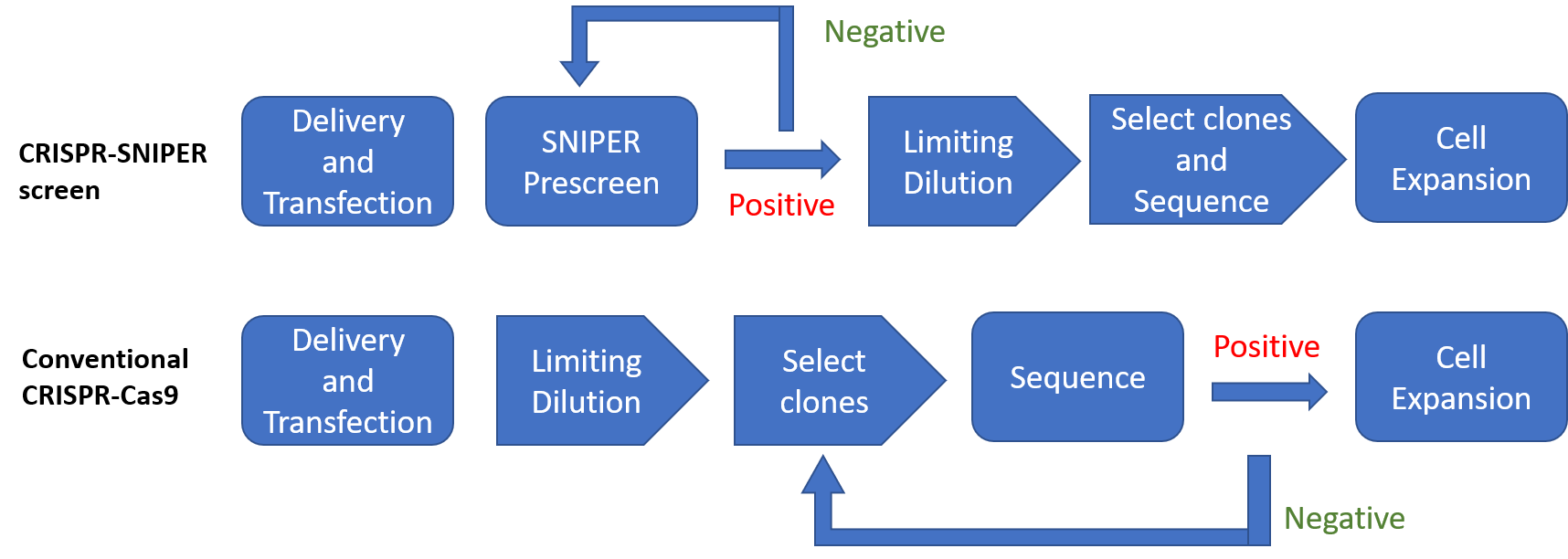
Figure 2: Conventional and REPROCELL SNIPER CRISPR-Cas9 workflow
SNIPER (Specification of Newly Integrated Position and Exclusion of Random-integration) combines a checkerboard of culture conditions with highly sensitive digital PCR to empirically pre-screen and identify clones most likely to be positive for the desired gene-edit. As a result, fewer clones are selected for checking, more positive clones are identified, and the screening process is significantly reduced from around 8 weeks to 2 weeks (Table 1, Figure 2).
| Screening | Average no. of clones selected | Positive clones (%) | Average Duration (weeks) |
| SNIPER | 50 | 20 - 30 | 1 - 2 |
| Conventional | >200 | 0.1 - 1 | 8 |
Table 1: Comparison of conventional and SNIPER screening workflows for a typical knock-in gene editing study
In addition to use as a screening and analytical tool, the addition of SNIPER to CRISPR-Cas systems can also be utilized to facilitate optimization steps required for the Design and Delivery stages of the technology. Component design, multiple component delivery and transfection strategies can be assessed under different culture conditions within the checkerboard to optimize and select the best combination for a cell type and gene-edit, negating the need for multiple optimization steps in the pre-analysis stages. Crucially, through the application of CRISPR-SNIPER, success or failure is identified at an early stage in the workflow, ensuring that time and costs are kept to a minimum for unsuccessful gene-editing attempts.
CRISPR-SNIPER technology is extremely versatile, maximizing gene-editing efficiencies in numerous different cell lines, including induced pluripotent stem cell (iPSC) lines. Due to the improved detection capability of SNIPER screening, fragile cell types more likely to be negatively affected by gene-editing procedures do better using CRISPR-SNIPER protocols.
As well as the technical advantages offered by the CRISPR-Cas9 system, the addition of the SNIPER screen accelerates the identification of successful editing for all knock-in, knock-out, and single nucleotide base-pair change applications.
Multiplex Gene Knock-out
Potent and precise knock-out (KO) of genes has traditionally been an important research tool for determining the function of a gene. CRISPR-SNIPER significantly streamlines the ‘loss of function’ process with elevated levels of specificity and efficiency. The addition of a SNIPER screen enables a multiplex approach to gene KO to be undertaken. Conventional CRISPR-Cas9 methods allow for the step-wise KO of multiple genes.
However, several culture passages in between each gene KO procedure are required, resulting in an extended experimental process that may adversely affect the viability and function of cells. This is a common issue for iPSC lines where extended passage can lead to decreased growth rates, formation of genetic abnormalities, and inhibition of differentiation capacity.
By using SNIPER technology, several genes can be targeted at once, thereby reducing the number of passages and the time required to obtain a viable multiple gene KO cell product (Figure 3).
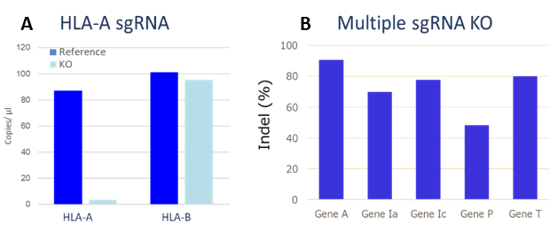
Figure 3: SNIPER knock-out gene editing. Panel A: Edits were generated using a sgRNA specific for the Human Leukocyte Antigen (HLA) class A gene. Selective and complete knock-down of the HLA-A gene was achieved by day 7 of culture. Panel B: 5 sgRNA constructs specific for each gene were included in one optimized experiment. INDELS for all 5 genes were detected.
Long-length DNA Knock-In
Specific knock-in (KI) of genes or ‘gain of function’ has many potential applications, from the addition of a reporter gene for live imaging of cells and identification or purification purposes to gene repair or replacement required for many potential gene therapy options.
Conventional CRISPR techniques are limited by the length of DNA that can be inserted and subsequently detected. The addition of SNIPER to CRISPR-Cas workflows allows for the accurate detection of both short and longer length DNA inserts incorporated using CRISPR-Cas9 and the HDR pathway (Figure 4).
Detection of longer length inserts using conventional CRISPR methods is often difficult due to the extended homology regions or arms of the insert. SNIPER screen utilizes a high-fidelity probe and digital PCR system to accurately detect the inserted DNA and enable detection of longer length inserts up to 2kb. The addition of the screen increases the potential use of CRISPR-Cas9 technology to applications where larger DNA insertions are required to obtain the edit of interest.
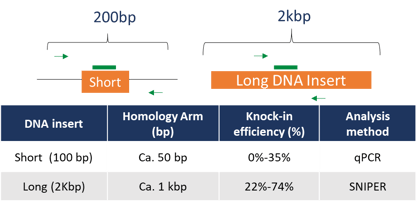
Figure 4: Comparison of a long and short sequence specific oligo donor in a typical knock-in gene editing study. Longer length homology arms inhibit reliable detection using conventional CRISPR techniques.
Single Nucleotide Base change applications
The largest class of known human pathogenic mutations, by far, is the single nucleotide polymorphism (SNP). Installing, changing, or reversing pathogenic SNPs efficiently and cleanly within a vast genome can be difficult using conventional CRISPR techniques. For single nucleotide base change generation, the use of a traditional screening gene, such as PuroR or GFP, to aid selection is not an attractive option.
Consequently, screening of large numbers of colonies is required to identify positive clones, resulting in a significant investment in time and resources. Addition of SNIPER to single base change modification screening results in a 100-fold or greater increase in the fraction of positive clones, greatly improving this resource-intensive step. This increase in efficiency is crucial to reliably differentiate between colonies heterozygous or homozygous for a SNP (Figure 5).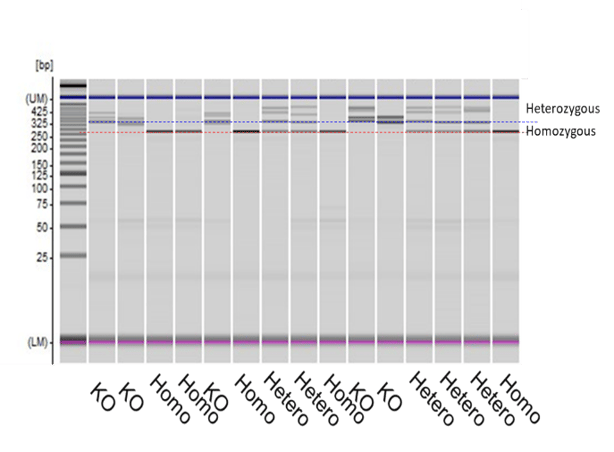 Figure 5: CRISPR-SNIPER Detection of SNP conversion in iPSC line. Knock in mutants were generated by inserting DNA containing the SNP of interest. SNIPER screen detected the number of colonies positive for either the homozygous (33%) or heterozygous (26%) version of the SNP. Wild type (KO) SNP colonies were also quantified (33%).
Figure 5: CRISPR-SNIPER Detection of SNP conversion in iPSC line. Knock in mutants were generated by inserting DNA containing the SNP of interest. SNIPER screen detected the number of colonies positive for either the homozygous (33%) or heterozygous (26%) version of the SNP. Wild type (KO) SNP colonies were also quantified (33%).
Disease modelling using CRISPR-SNIPER
Laboratory disease models are useful tools for investigating the mechanisms of disease. Obtaining disease-affected tissue from which to develop and maintain a disease model can often be difficult due to the rarity of a condition or the diseased state of a tissue.
Moreover, disease-causing mutations are varied and often complex, ranging from single gene changes through to multifactorial disorders, making the creation of a laboratory model challenging. CRISPR-Cas systems offer an opportunity to develop disease models from healthy tissue that are genetically representative of a disease. Whether a KO, KI or single nucleotide base change, CRISPR-SNIPER technology can reliably and efficiently generate cells that mimic disease characteristics within an experimentally brief time frame.
Figure 6 demonstrates the establishment of an iPSC-based disease neuronal cell model of Alzheimer’s disease (AD) using CRISPR-SNIPER to edit the AD-associated gene, PS1. Such models are comparable with patient-derived models and provide a valuable alternative to investigate disease pathogenesis.
Adding SNIPER screen to the CRISPR-Cas workflow therefore not only improves success for the most challenging gene-editing approaches, but significantly accelerates the optimization and experimental time frame, making the CRISPR-SNIPER system commercially transferrable, cost-effective, and user-friendly.
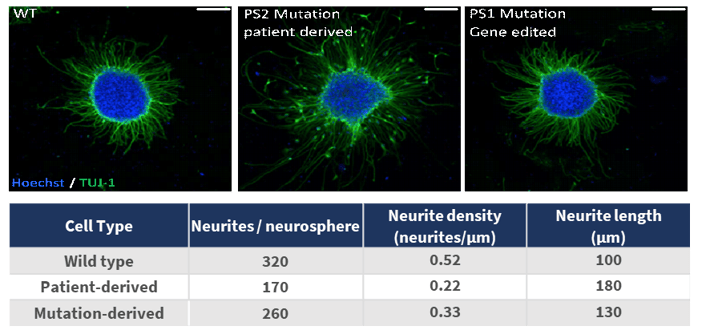
Figure 6. Creation of an Alzheimer’s Disease Model. Wild type (WT) iPSCs were generated from a healthy donor, and CRISPR-SNIPER gene editing used to introduce a mutation in the AD-associated gene, PS1. In parallel, an iPSC line bearing a mutation in the AD-associated gene, PS2, was generated from tissue donated from a donor with AD. Comparison of neurite characteristics demonstrated that neurospheres from iPSC lines with the different genetic backgrounds displayed quantifiable differences in neurite length and number.
REPROCELL CRISPR-SNIPER Gene-editing Services
Unlike comparable gene-editing companies and providers, REPROCELL offers a modular and flexible CRISPR-SNIPER approach that benefits many different and complex gene-editing needs including the experimentally challenging constructions such as marker insertions and single nucleotide base changes (Figure 7). Importantly, the modules are applicable to a variety of cell types, most notably iPSCs and both adherent and suspension cancer cells.
Module 1: Design and Feasibility Package
This module enables quick and efficient optimization of individual components, component delivery and culture conditions for a specific gene-edit and cell type.
REPROCELL request only the sequence and locus of your target gene with no additional methodology required. In-house expert scientists will custom design and manufacture the sgRNA and donor DNA fragments for your experiment and will test up to 6 transfection conditions to identify the best component design and delivery condition with the highest transfection efficiency.
The end point is a bulk unselected clone stock ready for individual clone-selection, clone-characterization, and clone-expansion.
Module 2: Full-Service Package
Module 2 offers a more comprehensive and extensive approach. In addition to the unique provisions of module 1, clone-selection, clone-characterization, and clone-expansion elements are incorporated into the module to provide a full optimization and gene-editing service. The end point for module 2 is the provision of individual selected and expanded clones with the gene-edit of choice.
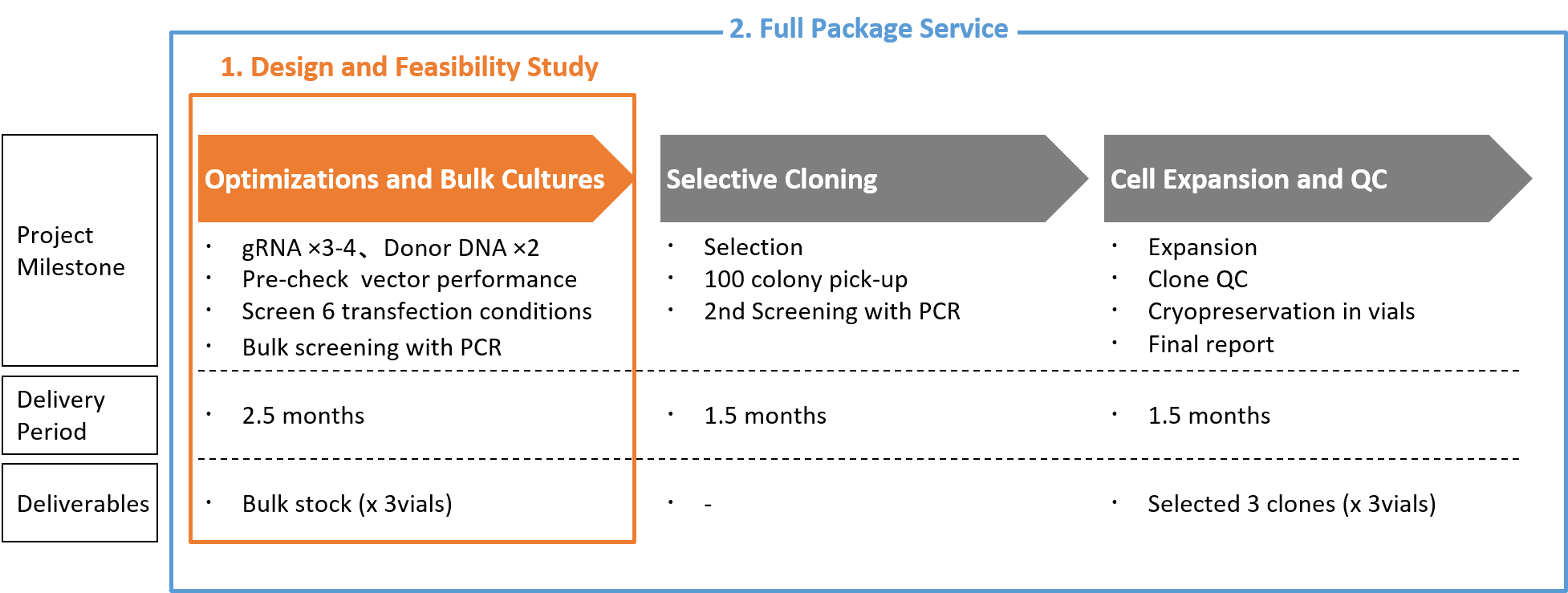
Figure 7: With CRISPR-SNIPER, Two types of service menus are offered: (1.) Design and Feasibility study and (2.) Full package service. The process of Design and Feasibility study is our proprietary technology and know-how to achieve success rate as high as 95% even for SNP conversion.
References
- Mojica et al. Intervening Sequences of Regularly Spaced Prokaryotic Repeats Derive from Foreign Genetic Elements. Journal Molecular Evolution 60 (2005)
- Deltcheva et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471 (2011)
- Gasiunas et al. Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. PNAS 109 (2012)
- Cong et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339 (2013)
- You et al . Advancements and Obstacles of CRISPR-Cas9 Technology in Translational Research. Molecular Therapy Methods & Clinical Development 13 (2019)
- Clarke et al. A robust and reproducible human pluripotent stem cell derived model of neurite outgrowth in a three-dimensional culture system and its application to study neurite inhibition. Neurochemistry International 106 (2017)
Note: The modified cells are developed, manufactured or supplied by GenAhead Bio Inc. under license from ERS Genomics Limited.







