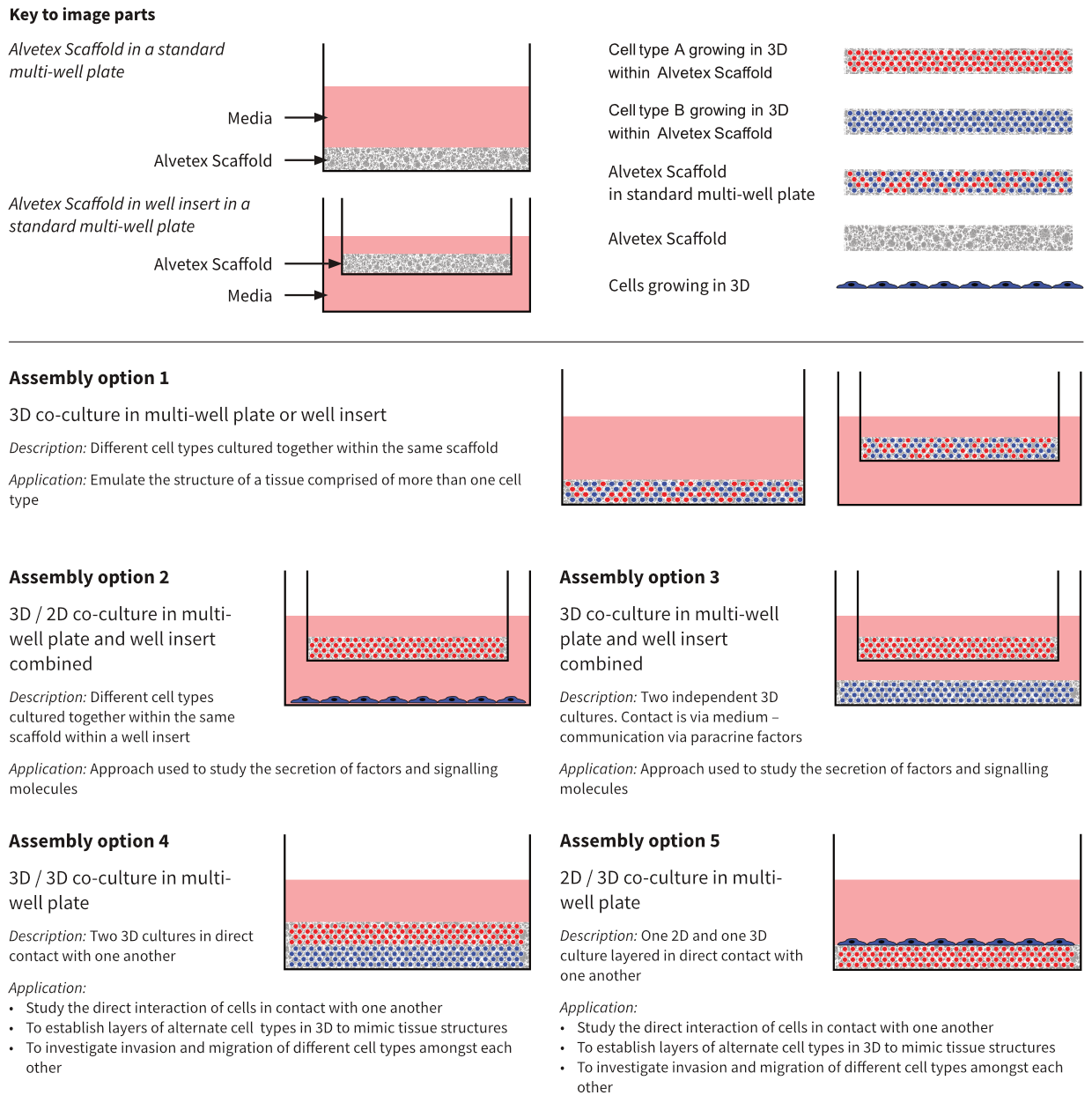
Figure 1: Alternative approaches for co-culture design using Alvetex Scaffold. Alvetex Scaffold is a versatile technology that enables users to create co-culture models in many different ways. These schematics illustrate some of the potential options available.
Example Co-culture Models
1. Development of Glial and Neuron Co-Cultures to Model Neural Tissues
Understanding the complex interactions between different cell types in the brain is important for elucidating how the brain functions, as well as for developing therapeutic approaches to combat disease states. Neurons are specialised cells for conducting nerve impulses and are supported and protected by glial cells. Culturing neurons and glial cells together enables the two-way communication between these cells to be studied. Using Alvetex Scaffold to create a three dimensional model allows a more physiologically relevant setup for mimicking the in vivo functionality of both healthy and injured or diseased cells. These 3D models are reproducible, robust, and are more representative for example, of the formation and function of the glial scar in vivo after CNS injury. In this example cocultures of glial cells (U118-MG) and stem cell-derived neurons were set up to study cellular interactions and the effect of glial cells on neuritogenesis. More in-depth analysis of this co-culture can be found within the application note regarding neural tissues at Application Notes and Whitepapers.
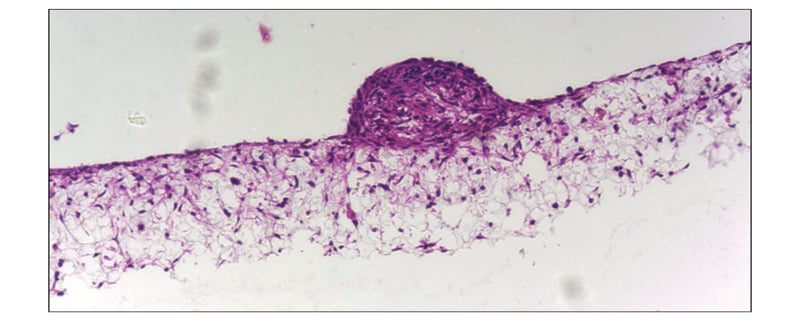 Figure 2: Co-culture of glial and neural cells to model brain tissues. Brightfield micrograph showing the structure of a human stem cell derived neurosphere co-cultured for 7 days with U118-MG glial cells on Alvetex Scaffold presented in the 12-well insert in 12-well plate format. Cells were fixed, embedded in paraffin wax, sectioned (10 μm) and counter-stained with haematoxylin and eosin. Scale bar: 200 μm.
Figure 2: Co-culture of glial and neural cells to model brain tissues. Brightfield micrograph showing the structure of a human stem cell derived neurosphere co-cultured for 7 days with U118-MG glial cells on Alvetex Scaffold presented in the 12-well insert in 12-well plate format. Cells were fixed, embedded in paraffin wax, sectioned (10 μm) and counter-stained with haematoxylin and eosin. Scale bar: 200 μm.
2. Model of Colon Cancer Cell Invasion Into the Underlying Stroma
Co-cultures of cancer cells and their surrounding stromal tissues are useful for the study of cancer cell invasion. The spread and invasion of cancer cells within the body is the most life-threatening aspect of the disease; therefore understanding the cellular and molecular mechanisms is indispensable for developing anti-metastatic drugs. The use of in vitro models to study cancer cell biology is preferred for both ethical and economical reasons. The tumour stroma is thought to influence the invasive potential of cancer cells [4], therefore by culturing cancer cells together with fibroblastsbin 3D, a more in vivo-like microenvironment can be created. In the example in Figure 3, the invasion of SW480 colon carcinoma cells into the fibroblast-filled scaffold demonstrates the highly invasive phenotype of these cancer cells.
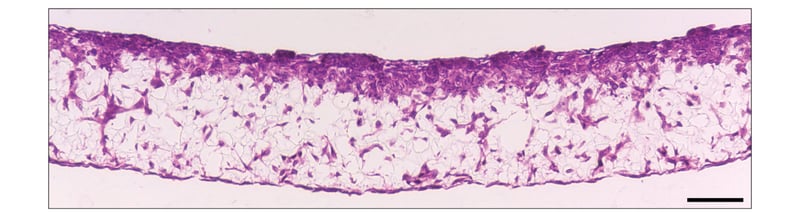 Figure 3: SW480 colon cancer cell invasion into Alvetex Scaffold containing 3T3 fibroblasts. Brightfield micrograph showing the structure of SW480 colon adeno-carcinoma cells co-cultured for 7 days with established 3D cultures of 3T3 fibroblasts on Alvetex Scaffold. Cultures were set up in 6-well inserts in the well insert holder in a deep Petri dish format. Cells were fixed, embedded in paraffin wax, sectioned (10 μm) and counterstained with haematoxylin and eosin. Scale bar: 100 μm.
Figure 3: SW480 colon cancer cell invasion into Alvetex Scaffold containing 3T3 fibroblasts. Brightfield micrograph showing the structure of SW480 colon adeno-carcinoma cells co-cultured for 7 days with established 3D cultures of 3T3 fibroblasts on Alvetex Scaffold. Cultures were set up in 6-well inserts in the well insert holder in a deep Petri dish format. Cells were fixed, embedded in paraffin wax, sectioned (10 μm) and counterstained with haematoxylin and eosin. Scale bar: 100 μm.
3. Full Thickness Human Skin Model
The development of in vitro full thickness skin models is useful for researching the major functions of the skin, investigating the potential toxicity of substances to human skin, as well as evaluating transdermal pharmaceuticals, thereby providing alternative methods to animal testing. The simple set-up and maintenance of cell cultures on Alvetex Scaffold enables the development of such complex organotypic models. The high porosity of the material increases the lifespan and value of the skin construct, and compatibility with a broad range of analytical techniques enables the user to simply and routinely produce dermal equivalents. In the example in Figure 4, dermal fibroblasts were cultured within the scaffold and the keratinocytes seeded on top differentiated to form stratified cell layers.
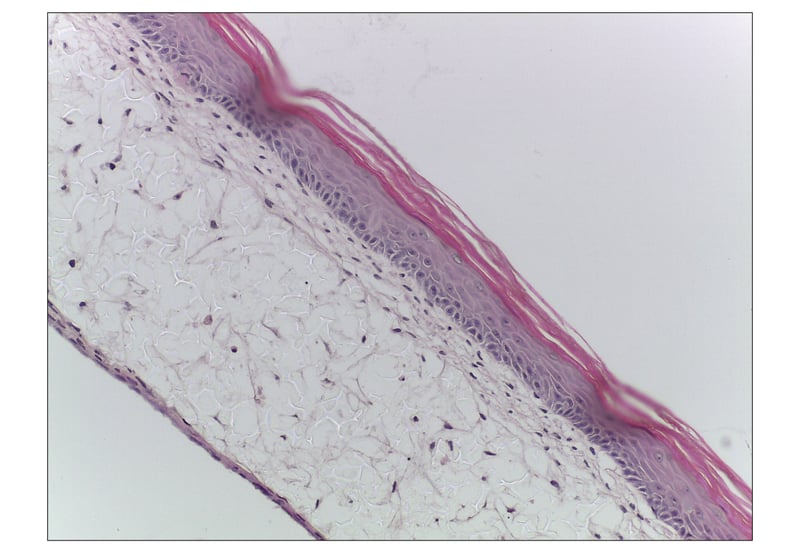 Figure 4: Fully-developed primary human skin equivalent using Alvetex Scaffold. Brightfield micrograph of the development of a human skin equivalent using primary dermal fibroblasts and primary human keratinocytes in Alvetex Scaffold 12-well insert format (AVP005) presented in a 6-well plate. After culturing for one month at the air-liquid interface, images show a human skin construct with an organised epidermis. Cell flattening within the stratum granulosum and several cornified layers lifting off the top surface of the construct can be observed. Viable dermal fibroblast growth can also be seen within Alvetex Scaffold. Cells were fixed, embedded in paraffin wax, sectioned (10 μm) and counterstained with haematoxylin and eosin. Scale bar: 50 μm.
Figure 4: Fully-developed primary human skin equivalent using Alvetex Scaffold. Brightfield micrograph of the development of a human skin equivalent using primary dermal fibroblasts and primary human keratinocytes in Alvetex Scaffold 12-well insert format (AVP005) presented in a 6-well plate. After culturing for one month at the air-liquid interface, images show a human skin construct with an organised epidermis. Cell flattening within the stratum granulosum and several cornified layers lifting off the top surface of the construct can be observed. Viable dermal fibroblast growth can also be seen within Alvetex Scaffold. Cells were fixed, embedded in paraffin wax, sectioned (10 μm) and counterstained with haematoxylin and eosin. Scale bar: 50 μm.
4. Modelling Simple Epithelia Using Alvetex Scaffold
Simple epithelia normally line the surfaces of organs throughout the body, and mostly form monolayers. Each cell usually forms dynamic interactions with neighbouring cells and is in direct contact with the underlying basement membrane, through which nutrients are absorbed and filtration occurs [5]. Growing epithelial cells in conventional 2D cell culture can therefore limit their morphology and behaviour. Alvetex Scaffold provides a platform for supporting the 3D cell culture of epithelial cells on top of a layer of extracellular matrix protein with a co-culture of underlying fibroblasts, thereby mimicking the lamina propria and sub-mucosal tissues found beneath the basement membrane of epithelial cells in vivo.
Owing to its extremely high porosity, Alvetex Scaffold provides a greater opportunity for diffusion and exchange of solutes and ions beneath the epithelia than existing Transwell™ inserts. In addition, growing epithelial cells on a thin layer of extracellular matrix protein is more physiologically relevant than Transwell™ models. The culture of epithelial cells on Alvetex Scaffold enables new opportunities for the study of epithelial function, ion transport assays and drug absorbance studies, as well as their interaction with other cell types and modelling pathological conditions. In the example in Figure 5, Caco-2 colorectal adenocarcinoma cells were cultured on collagen 1 coated Alvetex Scaffold containing CCD-18Co colon fibroblasts, to mimic the in vivo intestinal mucosa.
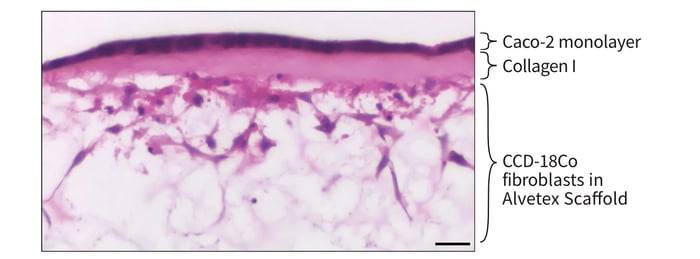 Figure 5: Co-culture of Caco-2 cells and CCD-18Co fibroblasts separated by a collagen I layer in Alvetex Scaffold. High magnification brightfield micrograph of an even monolayer of Caco-2 cells on the collagen-coated top surface of Alvetex Scaffold, with underlying CCD-18Co fibroblasts within Alvetex Scaffold. CCD-18Co fibroblasts were grown on Alvetex Scaffold presented in 6-well inserts in a 6-well plate format for 2 days prior to layering with Collagen I and seeding Caco-2 cells. Co-cultures were grown for a further 5 days, after which they were fixed, embedded in paraffin wax, sectioned (10 μm) and counter-stained with haematoxylin and eosin. Scale bar: 25 μm.
Figure 5: Co-culture of Caco-2 cells and CCD-18Co fibroblasts separated by a collagen I layer in Alvetex Scaffold. High magnification brightfield micrograph of an even monolayer of Caco-2 cells on the collagen-coated top surface of Alvetex Scaffold, with underlying CCD-18Co fibroblasts within Alvetex Scaffold. CCD-18Co fibroblasts were grown on Alvetex Scaffold presented in 6-well inserts in a 6-well plate format for 2 days prior to layering with Collagen I and seeding Caco-2 cells. Co-cultures were grown for a further 5 days, after which they were fixed, embedded in paraffin wax, sectioned (10 μm) and counter-stained with haematoxylin and eosin. Scale bar: 25 μm.
5. Demonstration of Co-Culture Enhancing Cell Function
In this example, the function of human upcyte® hepatocytes (Medicyte) was assessed in conventional 2D culture and 3D culture using Alvetex Scaffold. In 3D culture, the activity of the upcyte® hepatocytes was tested alone or in co-culture with microvascular endothelial cells. Figure 6 shows data for the activity of CYP3A4 in upcyte® hepatocytes cultured on a 2D plate and on Alvetex Scaffold presented in both a 12-well plate and 6-well insert formats. Hepatocytes were grown as monocultures and also co-cultured with upcyte® microvascular endothelial cells for 10 days. Culturing the cells in 3D on Alvetex dramatically increased CYP3A4 activity, an effect that was substantially improved by co-culture with endothelial cells, but only when cultured in 3D. This indicates that the 3D environment provided by Alvetex Scaffold was essential for forming beneficial cell-cell interactions between the hepatocytes and endothelial cells.
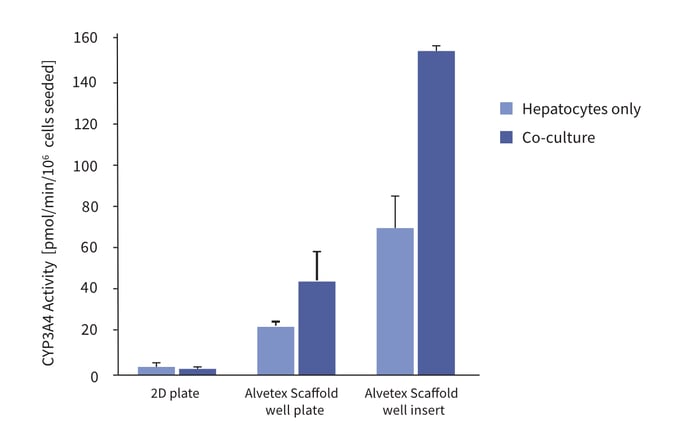 Figure 6: Enhanced cell function with hepatocyte and endothelial cell co-culture. The activity of CYP3A4 in upcyte® hepatocytes cultured on a 2D plate and on Alvetex Scaffold presented in both a 12-well plate and 6-well insert formats. Hepatocytes were grown as monocultures and also co-cultured with upcyte® microvascular endothelial cells for 10 days.
Figure 6: Enhanced cell function with hepatocyte and endothelial cell co-culture. The activity of CYP3A4 in upcyte® hepatocytes cultured on a 2D plate and on Alvetex Scaffold presented in both a 12-well plate and 6-well insert formats. Hepatocytes were grown as monocultures and also co-cultured with upcyte® microvascular endothelial cells for 10 days.
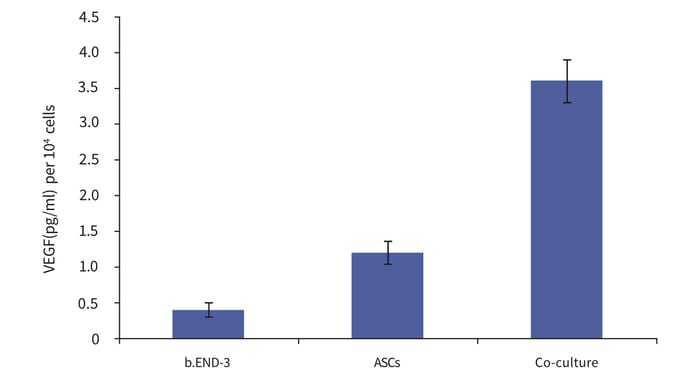 Figure 7: Co-culture of adipose tissue-derived stem cells with endothelial cells influences their differentiation. Adipose tissue-derived stem cells (ASCs) and endothelial cells (b.END-3) were cultured independently and as co-cultures in Alvetex Scaffold 12-well plate format for 3 days. Data from 3 sample replicates. VEGF levels normalised to the number of cells at the time of seeding, expressed as pg/mL per 106 cells. For further details please refer to Neofytou et al. 2011 [6].
Figure 7: Co-culture of adipose tissue-derived stem cells with endothelial cells influences their differentiation. Adipose tissue-derived stem cells (ASCs) and endothelial cells (b.END-3) were cultured independently and as co-cultures in Alvetex Scaffold 12-well plate format for 3 days. Data from 3 sample replicates. VEGF levels normalised to the number of cells at the time of seeding, expressed as pg/mL per 106 cells. For further details please refer to Neofytou et al. 2011 [6].
6. Co-culture Promotes Adipose Tissue-Derived Stem Cell Differentiation into a Pro-Angiogenic Phenotype
This model shows how 3D co-culture can influence the differentiation of cells. Adipose tissue-derived stem cells (ASCs) and endothelial cells (b.END-3) were cultured independently and as co-cultures in Alvetex Scaffold 12-well plate format for 3 days. Elevated levels of the angiogenic cytokine VEGF were found in conditioned media harvested from co-cultures compared to controls, indicative of a proangiogenic phenotype.
Conclusions
Alvetex Scaffold provides a versatile platform that enables cells to maintain their natural 3D shape and organisation, thereby allowing the investigation of more in vivo-like cell behaviour and function than with conventional 2D model systems. The ability to grow more than one cell type simultaneously enables the study of complex intercellular interactions by more accurately simulating tissue microenvironments. These co-culture experiments can be valuable for understanding and observing cell behaviour and differentiation, for studying cell signalling and migration dynamics, and for recapitulating tissues in both health and diseased states, with the potential for developing novel therapeutics. Alvetex Scaffold is versatile and simple to use, enabling the routine generation of genuine 3D cell cocultures. Available in a variety of formats, Alvetex Scaffold can be used in multiple set-up permutations, potentially including high-throughput applications.
References
- Bhandari RNB, et al. Liver Tissue Engineering: A Role for Co-culture Systems in Modifying Hepatocyte Function and Viability. Tissue Engineering 2001; 7(3): 345-357.
- Miki Y, et al. The advantages of co-culture over mono cell culture in simulating in vivo environment. J Steroid Biochem Mol Biol 2012; 131(3-5): 68-75.
- Truskey GA. Endothelial Cell Vascular Smooth Muscle Cell Co-Culture Assay For High Throughput Screening Assays For Discovery of Anti-Angiogenesis Agents and Other Therapeutic Molecules. Int J High Throughput Screen 2010; 1: 171–181.
- Kramer N, et al. In vitro cell migration and invasion assays. Mutat Res: Rev Mutat Res 2012. http://dx.doi.org/10.1016/j.mrrev.2012.08.001.
- Hagios C, et al. Tissue architecture: the ultimate regulator of epithelial function? Philos Trans R Soc Lond B Biol Sci 1998; 353: 857-870.
- Neofytou EA, et al. Adipose tissue-derived stem cells display a proangiogenic phenotype on 3D scaffolds. J Biomed Mater Res A 2011; 98(3): 383-93.
Appendix 1: Methods
Method 1: Development of Glial and Neuron Co-cultures to Model Neural Tissues
Neuroprogenitor aggregate production
TERA2.cl.SP12 embryonal carcinoma stem cells were maintained at a high density in an undifferentiated state. 1.5 × 106 of these cells were seeded in an untreated Petri dish in 20 ml of medium and left to aggregate overnight. 0.1 μm EC23 retinoid (REPROCELL) was added (or alternatively 10 μm ATRA/AH61), and gently swirled to distribute. Cells were left to differentiate for 21 days, with media changes every 3-4 days.
Medium: Dulbeccos Modified Eagles Medium (DMEM) supplemented with 10 % v/v heat-treated FBS, 2 mM L-glutamine and 100 U/ml Penicillin/Streptomycin.
Medium change method: Medium and neurospheres were transferred to a 50ml tube and left to stand for 20 minutes to let the neurospheres settle. The medium was then aspirated and replaced, (including the retinoid differentiation compound). The suspension was gently mixed once with a 10 ml pipette and poured into a fresh (untreated) Petri dish.
Inducing neurite outgrowth
One day before induction: Alvetex Scaffold 12-well inserts presented in a 12-well plate were prepared for seeding with a 70 % ethanol wash (4 ml per well) and subsequent media washes (twice with 4 ml). 1.5 ml 10 μg/ml of laminin and Poly-D- Lysine in PBS was added to each insert. The plate was then sealed with Parafilm and left overnight.
After a minimum of 21 days of differentiation: Using a 100 μm cell strainer, the neurospheres were filtered and then back filtered into 10 ml of fresh media without any retinoid compound. 1 × 106 U-118MG glioblastoma cells were seeded into the centre of the Alvetex Scaffold insert in 100 μl and left to settle for 15 minutes. 200 μl of media and between 6-10 neurospheres were removed from the medium and placed onto the centre of the polymer. Outgrowth culture medium containing the following mitotic inhibitors was prepared: 1 μM cytosine arabin-oside, 10 μM, 5, fluoro-2-deoxyuridine, 10 μM Uracil. 2 ml was added to each well of the 12 well plate. Neurospheres were cultured for 7-10 days without any media changes. The media was slowly aspirated from the wells using a 1ml pipette and discs were fixed with 4 % paraform-aldehyde for 1 day and then washed 3 times with PBS. Samples were embedded and stained using protocols available at Alvetex Protocols.
Method 2: Model of Colon Cancer Cell Invasion into the Underlying Stroma
Alvetex Scaffold 6-well inserts presented in a 6-well plate were prepared for seeding with a 70 % ethanol wash (10 ml per well) and subsequent media washes (twice with 10 ml). 3T3 fibroblasts (ATCC, CCL-92) were seeded at a density of 0.5 × 106 cells in 50 μl per insert. The plate was incubated for a minimum of 15 minutes at 37°C with 5 % CO2 to allow the cells to settle into the scaffold. 3T3 medium was added to each well to a total volume of 10 ml taking care not to dislodge cells from Alvetex Scaffold. Plates were re-incubated and maintained for a further 7 days by complete 3T3 media exchange after every 2-3 days.
After 7 days, fibroblast 3D cultures were transferred to deep Petri dish holders and dishes were filled from the outside of the insert with enough SW480 medium to allow the medium to rise inside the insert and cover the substrate, but not to go over the sides of the inserts, i.e. 50ml ± 5 ml, as described for “feeding above and below separately” in the Quick Start protocol. SW480 cells were seeded at a density of 1 × 106 cells in 100 μl of medium per insert. The Petri dish was incubated overnight at 37°C with 5 % CO2 to allow the cells volume of 70 ml taking care not to dislodge cells from Alvetex Scaffold. Co-cultures were maintained for a further 7 days and the culture medium changed every 2-3 days.
Cultures were subsequently fixed and processed for histology and H&E counter-staining.
3T3 medium: DMEM High glucose supplemented with 10% v/v FBS, 2μM L-glutamine and 100 U/ml Penicillin/Streptomycin.
SW480 medium: DMEM High glucose supplemented with 10 % v/v heat-inactivated FBS, 2 μM L-glutamine and 100 U/ml Penicillin/Streptomycin.
Method 3: Full Thickness Human Skin Model
Alvetex Scaffold 12-well inserts presented in a 6-well plate were prepared for seeding with a 70 % ethanol wash (10 ml per well) and subsequent media washes (twice with 10 ml of medium 1). NB. Refer below for all media formulations. Primary fibroblasts isolated from human foreskin (P5) were cultured on the inserts for 1 week: 1 × 106 fibroblasts were seeded in 100 μl of culture medium 1 per insert and incubated for 1 hr at 37°C with 5 % CO2. The inserts were then submerged in culture medium 1 (10.5 ml) for 7 days, with media 2 changes every other day.
After 7 days of fibroblast culture, culture medium 1 was removed from the wells. Primary human keratinocytes (0.5 × 106 cells) isolated from foreskin (P4) were seeded on to the inserts in 100 μl of culture medium 2, and incubated for 1 hr at 37°C with 5 % CO2. After 1 hour the inner chamber of the well insert to settle on top of the scaffold. The following morning SW480 media was added to each Petri dish to a total was filled with culture medium 2 (2 ml) and the outer well compartment (i.e. underneath the insert) was filled with 5 ml of culture medium 1.
After 3 days all culture medium was removed and 4 ml of culture medium 3 was added beneath the well inserts in order to maintain the cultures at the air/liquid interface. Culture medium 3 was changed every other day. The cultures were maintained at the air/liquid interface for 1 month and the Alvetex Scaffold discs then placed in the appropriate fixative for analysis. Cultures were then processed for histological analysis.
Medium 1 (fibroblast medium): DMEM (High glucose, PAA, E15-810) with 10 % v/v FBS and 100 U/ml Penicillin/Streptomycin.
Medium 2 (Keratinocyte proliferation medium, sub-merged conditions): Epilife (Invitrogen, M-EPI-500-CA) with human keratinocyte growth supplement (HKGS, Epilife, S-001-5) and 100 U/ml Penicillin/Streptomycin.
Medium 3 (Keratinocyte differentiation medium, feeding from beneath insert only): DMEM/ Ham’s F-1 2 (1:1) (PAA, E15-813) with Cholera toxin, (Sigma, C8052-2mg), final concentration 10–10 M; Epidermal Growth Factor (Mouse), (Serotec, EGF-1), final concentration 10 ng/ml; Hydrocortisone, (Sigma, H4881), final concentration 0.4 μg/ml; Insulin (Sigma, I5500), final concentration 5 μg/ml; Transferrin (Sigma, T2252-500mg), final concentration 5 μg/ml; 3,3ʹ,5-Triiodo-L-thyronine sodium salt, (Sigma T6397-100mg), final concentration 2 × 10–11 M; 10 % v/v FBS and 100 U/ml Penicillin/Streptomycin.
Method 4: Modelling Simple Epithelia Using Alvetex Scaffold
Alvetex Scaffold 6-well inserts presented in a 6-well plate were prepared for seeding with a 70 % ethanol wash (10 ml per well) and subsequent media washes (twice with 10 ml). CCD-18Co fibroblasts (ATCC, CRL-1459) were seeded at a density of 1 × 106 cells in 200 μl per insert. Enhanced CCD-18Co medium was added to each well to a total volume of 7 ml taking care not to dislodge cells from Alvetex Scaffold, i.e. feeding separately from above and below. Plates were re-incubated and maintained for a further 2 days.
After 2 days, medium was removed down to the air-liquid interface, i.e. feeding from below only, and 300 μl of 2 mg/ml rat tail collagen I prepared as per manufacturer’s instructions (BD Biosciences, cat # 354236) was added to the surface of each insert. Plates were re-incubated for 3 hours to allow the collagen gel to set, after which fresh medium was added up to 10 ml per insert and plates were re-incubated overnight.
The following day, medium was removed down to the air-liquid-interface and Caco-2 cells (ATCC, HTB-37), which had been cultured in enhanced CCD-18Co medium for at least two passages on 2D, were seeded at a density of 0.5 × 106 cells in 1ml per insert over the surface of the collagen gel. Plates were re-incubated overnight to allow cells to attach, after which fresh medium was added up to 10 ml. Co-cultures were maintained for a further 5 days and the culture medium changed every 2 days. Cultures were subsequently fixed and processed for histology and H&E counter-staining.
Enhanced CCD-18Co medium: High glucose (4.5 g/L) DMEM supplemented with 10 % v/v heat-inactivated FBS, 0.1 mM non-essential amino acids, 2 μM L-glutamine and 100 U/ml Penicillin/Streptomycin.
Detailed protocols for collagen I gel layering, histology and H&E staining are available on Alvetex Protocols.