Histology (2): Processing Alvetex® Scaffold 3D Cultures for PFA Fixation and Paraffin Wax Embedding for Immunofluorescence
● Download this protocol as a PDF (1.8 MB)
1. Introduction
Immunofluorescence uses the recognition of cellular targets by fluorescent dyes or antigen-specific antibodies coupled to fluorophores. Depending on the antibody or dye used, proteins, lipids and DNA can be visualised within individual cells and tissues. Alvetex Scaffold can easily be processed like a standard tissue sample, allowing established immunofluorescence methods to be followed with excellent results. An example protocol is outlined below.
2. Materials and Methods
2.1. Materials Required for Fixing, Embedding and Sectioning
- % Paraformaldehyde (PFA) fixative (For full method refer to Histology (1): Choosing the Right Fixative to Preserve 3D Cell Cultures.)
- Dehydration ethanols (30 %, 50 %, 70 %, 80 %, 90 %, 95 %, 100 %)
- Histoclear
Note: the use of Histoclear is recommended over the use of Xylene.
- Paraffin wax
- Basic equipment: spade forceps to handle 3D Alvetex Scaffold cultures, scalpel (to trim/ cut scaffold if required), disposable pipettes, embedding moulds, convection oven, microtome
2.2. Materials Required for Immunofluorescence
- 1× citrate buffer pH 6.0
- Blocking buffer (5 % Normal goat serum; 1 % bovine serum albumin; 0.2 % Triton X-100 in PBS)
- Permeabilisation solution (1 % w/v Triton X-100 in PBS)
- Primary and secondary antibodies (specific to experimental analysis)
- Microwave oven (if antigen retrieval is required)
- Humidified chamber
- Vectashield/ DAPI mountant
2.3. 4 % PFA Fixation of Scaffolds
- Aspirate off the medium and carefully wash the 3D culture in PBS twice.
- If using well inserts in Petri dishes, remove inserts from holder and place in a conventional 6 well plate. To fix, add in 5 mL 4 % PFA fixative at 4 °C. Fix specimens at 4 °C for a minimum of 12 hours but no longer than 24 hours.
- Aspirate off the fixative, wash 3 times using 5 mL PBS to thoroughly remove excess fixative, discarding the waste liquid.
- If using inserts, remove Alvetex Scaffold from the well insert either by separating the two parts of the device or using a scalpel to cut out the scaffold. At this time samples can be transferred to a tissue processing cassette to minimize direct handling and damage to the 3D culture.
- Aspirate off the PBS and add 5 mL of 30 % ethanol. Leave to equilibrate for at least 15 minutes. Aspirate off the ethanol and discard.
- Repeat with 50 %, 70 %, 80 %, 90 % and then 95 % ethanol. A gradual dehydration of the sample will result in less tissue shrinkage. Material can be stored in 95 % ethanol prior to paraffin embedding.
2.4. Paraffin Embedding and Sectioning
- Fully dehydrate specimens stored in 95 % ethanol by replacing with 100 % absolute ethanol for at least 30 minutes.
- Aspirate off the ethanol and replace with Histoclear for at least 30 minutes.
Note: Histoclear will dissolve certain types of plastic, therefore it is best to process samples in glass tissue processing cassettes.
- Replace the Histoclear with a 50:50 solution of Histoclear and molten paraffin wax (60 °C) mix and incubate in a convection oven at 60 °C for 30 minutes.
- Replace the Histoclear:wax mix with 100 % molten wax and incubate at 60 °C for a further 60 minutes.
- Transfer the scaffold to plastic embedding moulds and orientate into the required embedding position, with plane of section in mind. Embed in molten wax.
- Allow wax to cool and set at room temperature for 1-2 hours or overnight.
- Once the was has hardened, remove the wax embedded block from the plastic mould. The sample is now ready for sectioning on a suiltable microtome (e.g. Leica RM2125).
- Following the microtome manufacturers’ instructions throughout, align the block correctly with the microtome blade and proceed to cut 10 μm sections of the sample block.
- Transfer sections to a slide water bath (40 °C), floating them on the surface of the water to enable them to flatten out.
- Transfer selected sections to slides by flotation. Superfrost Plus slides (Thermo, 4951PLUS4) are recommended since sections adhere to these slides well.
- Place on a slide drier and leave overnight. The sections should now be ready for immunofluorescence.
2.5. Immunofluorescent Analysis
- Deparaffinise sections in Histoclear for 10 minutes. Handle samples carefully to avoid loss of section from slide.
- Hydrate specimen through a graded series of ethanols (100 %, 90 %, 70 %) with 5 minutes incubation in each solution.
- If antigen retrieval is required, place slides in 1x citrate buffer (pH 6) and microwave (800 W) for 6 minutes. If antigen retrieval is not required go to point 5.6.
- Leave to stand outside microwave for 1 minute then microwave for a further 3 minutes at 800 W.
- Allow to cool for 20 minutes and wash in PBS for 10 minutes. Repeat three times.
- Treat cells with permeabilisation solution for 15 minutes.
- Aspirate off permeabilisation solution, replace with blocking buffer and incubate for 15 minutes.
- Incubate with primary antibody diluted in blocking buffer (this will be specific to the antigen chosen and will be used at a pre-determined concentration) overnight at 4 °C in a humidified chamber, or as stated in the datasheet for the specific antibody used.
- Wash 3 times for 10 minutes in PBS.
- Incubate with secondary antibody diluted in blocking buffer (this will be specific to the antigen chosen and will be used at a pre-determined concentration) for 2 hours in the dark at room temperature.
- Wash 3 times for 10 minutes with PBS.
- Mount in Vectashield/ DAPI. This solution simultaneously mounts the specimen and stains cell nuclei.
- Seal the perimeter of the coverslip with nail varnish and dry in the dark.
- Store slides in the dark at 4 °C until ready for inspection using a fluorescence microscope equipped with the appropriate filters.
3. Example Results
Figure 1 below shows an example of a 3D Alvetex Scaffold culture fixed with 4 % PFA, embedded in paraffin wax, sectioned and probed with Ki67 antibody using immunocytochemistry and standard fluorescence microscopy.
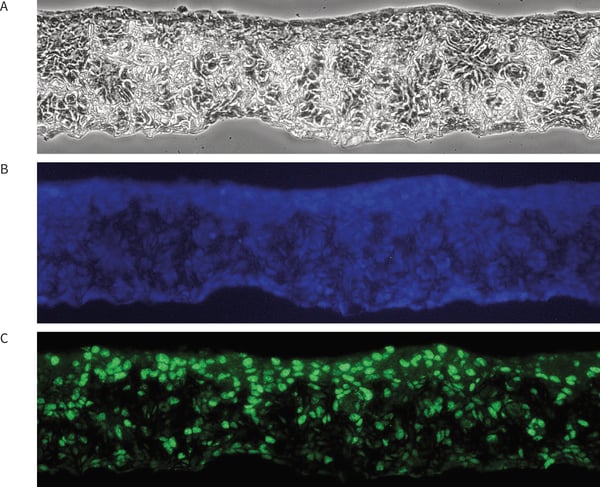
Figure 1. Protein expression in 3D cell cultures can be localised using immunecytochemistry and fluorescence microscopy. Three corresponding images from the same region of Alvetex Scaffold show: (A) cell morphology by phase microscopy; (B) expression of cell nucleus marker DAPI; (C) expression of cell proliferation marker Ki67.