Alvetex Strata Application Note 01
Maintenance of Intact Tissues Using Alvetex Strata
Download this application note as a PDF (6.92 MB)
Overview
REPROCELL has developed Alvetex® Strata, a highly porous membrane presented in a well insert format. This product has multiple applications, including the ability to stably support intact viable tissues during cell culture. Here we demonstrate two novel applications for this new technology: (1.) its use for the stable attachment of neural tissue during live cell imaging; (2.) the maintenance of human embryonic stem cell-derived embryoid bodies as intact three dimensional tissues and their subsequent analysis. Alvetex Strata is a new product and has distinct advantages over existing well insert technologies using semi-porous membranes, notably:
- Enhanced porosity for improved nutritional support from the medium;
- Modified surface topography to improved tissue attachment;
- Versatility for co-culture and construction of advanced in vitro models.
Introduction
REPROCELL specializes in the development of enabling technology to enhance the growth, differentiation and function of cultured cells and tissues. Our Alvetex products are designed to enhance the physiological relevance of in vitro models, provide researchers with new opportunities to culturing and studying cells and tissues in the laboratory, and improve methods for sample handling and analysis of cell-based assays.
Alvetex Scaffold is our market leading product that is primarily used for three-dimensional culture of dissoc-iated mammalian cells within the scaffold. In this article we introduce Alvetex Strata, which is our second generation of porous material, primarily designed to support the growth of cells and intact tissues on the surface of the membrane. At first glance, the structure of Alvetex Strata appears similar to Alvetex Scaffold: both products are made from the same crossed-linked polystyrene; both membranes are 200 microns thick; both are presented in our custom 6-well and 12-well inserts; and both come blister packed, sterile, ready for use. The difference however between these two materials concerns their fine structure and architecture. Both materials are highly porous and comprised of voids and interconnecting pores: in Alvetex Strata the voids and pores are significantly smaller (average 13- and 5-micron diameter, respectively) compared to those in Alvetex Scaffold (see Figure 1).
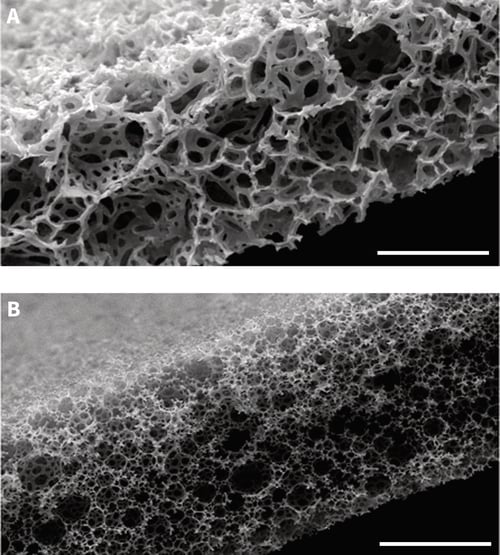
Figure 1. Scanning electron micrographs showing the structure of Alvetex Scaffold (A.) and Alvetex Strata (B.) porous polystyrene membranes in transverse section. Note that the size of the voids is significantly smaller in Alvetex Strata compared to Alvetex Scaffold. Scale bar: 100 microns.
In this application note, we demonstrate the use of Alvetex Strata to support the growth and maintenance of intact tissues and cell aggregates. Two example applications are provided, both of which offer alternative approaches and improvements of existing methodologies.
Example Application 1: Maintenance of Spinal Cord Organotypic Slices on Alvetex Strata
Background
The pioneering work of Ross Harrison (1907) in which axonal growth was first observed from fragments of frog neural tube into clotted lymph ultimately lead to the development of ‘cell culture’ and ‘tissue culture’ both of which have become obligatory tools in neurobiological research today.
The culture of dissociated cells was established when enzymatic methods such as use of trypsin were developed (Nakai, 1956). This has led onto the growth of cells as monolayers on flat transparent substrates and imaging of individual cells by phase microscopy. In the field of neural development, for example, this has been developed further and live cell imaging and the first observation of neurite outgrowth recorded (Banker and Cowan, 1977). However, this reductionist view is at the expense of cellular complexity normally found in real tissues and the introduction of artefacts and abnormal behaviours displayed by cells responding to changes in their micro-environment.
In contrast, such three-dimensional cyto-architecture is preserved in tissue culture where slices or fragments of brain tissue remain intact. ‘Organotypic slice cultures’ were originally developed in the 1940’s and allow tissue slices to retain their basic structural organization (Hogue 1947). Within such tissue slice models the occurrence of morphogenetic events such as cell proliferation, migration, differentiation and function of neurons can be assessed. Today, many modern molecular and cellular techniques have been developed to track the behavior of cells in tissue slices, including track tracing and dye labels, live cell imaging using fluorescent reporter molecules, genetic manipulation and reporters, electro-physiological recording, etc. (e.g. Gahwiler et al. 1997). Live cell imaging in particular, is a useful approach to study the proliferation and migration of neural cells within intact brain and spinal cord tissue slices (Miyata et al. 2005).
Modern methods have now been developed for the preparation and culture of precision-cut organ slices (Fisher and Vickers 2013; Gahwiler et al. 2001). Most often tissue slices are generated by mechanical sectioning techniques using a vibratome. Tissues are frequently maintained in culture on a porous support, most often suspended in a Petri dish or multi-welled plate. This method is originally based on the pioneering work of Luc Stoppini (1991), and is now used widely for tissue slice culture.
Millipore have developed a procedure for organotypic cell culture using their Millicell® product range incorporating a porous membrane housed in a culture plate insert. The membranes differ in material type (e.g. PTFE, polycarbonate, PET) and pore dimension. In all cases the surface of the membrane is flat and smooth and the porosity of the material is at best approximately 30%.
This is in contrast to Alvetex Strata which has a rough, topographical surface consisting of open voids and barbs onto which tissue slices can attach. Moreover, Alvetex Strata is 90% porous enabling enhanced exchange between the tissue slice and incubating medium, especially when using the Stoppini method of feeding.
We demonstrate herein that tissues remain viable and attach readily to the surface of Alvetex Strata without any additional coating required. Secure support of tissues is essential during live cell image recordings where the tissue slice must remain stationary with minimal movement.
Methodology
This is an example of a tissue slicing technique that enables the optimal preservation of the three-dimensional cytoarchitecture of the spinal cord. This is necessary to enable labeled cells to interact within a natural cellular environment. Such tissue slices can be kept stable and viable for weeks on Alvetex Strata membranes, allowing the proliferation, differentiation and migration of labeled endogenous radial glial cells to be observed over time.
This method provides a brief description of setting up organotypic spinal cord slice cultures to directly observe the behavior of labeled radial glia with a multi-photon time-lapse microscope.
- To prepare tissue slices, dissected rodent spinal cords were immediately immersed in ice cold medium and 400μm tissue slices were cut using a vibratome (Leica VT1200) and transferred to a Petri-dish.
- Tissue slices were then transferred to a class II laminar flow hood and placed on a porous membrane in a well insert. Two types of device were tested and compared: Millicell® well insert fitted with the Biopore™ (PTFE) membrane; REPROCELL well insert fitted the Alvetex Strata membrane.
- The well inserts were placed in a 6-well multi-welled plate with 1-5 ml of medium (64 % Dulbecco’s Modified Eagle Medium, 25 % HBSS, 10 % heat-inactivated fetal calf serum and 1 % Penicillin/Streptomycin) per well.
- Tissue slices were electroporated using a GFP expressing plasmid driven by a brain lipid binding protein (BLBP)-promoter; resulting in a portion of the radial glial cells in the ventricular zone of the spinal cord being labeled.
- The cultures were then placed in an incubator with 5 % CO2 at 37 °C overnight.
- GFP labeled cells were then imaged for up to 7 days in an incubation chamber using an Olympus multi-photon microscope (FV1000) (Figure 3).
- Images were captured at the beginning of the recording time and after regular periods to observe any differences in cellular positioning, growth, differentiation or migration.
Figure 2. Experimental set-up showing maintenance of tissue slice on Alvetex Strata and positioning of objective microscope lens.
Results
The data illustrates differences in the amount of tissue drift observed (while keeping a stable imaging field) when comparing the Millicell® and Alvetex Strata membranes after 24 hours of imaging recording. In the top two panels, the imaging field is shown at the start of imaging (A) and after 24 hours (B) using a Millicell® membrane. The Red arrow indicates the direction and extent (405 microns) of tissue drift. Likewise, the bottom panels show the identical conditions for a tissue slice maintained on an Alvetex Strata membrane (C,D). However, the amount of tissue drift (25 microns) was substantially reduced during the 24 hours period. This difference is largely due to the ability of the tissue to fully adhere to the Alvetex Strata membrane which has a rough three-dimensional surface topography. In comparison, the Millicell® membrane is smooth and flat.
Tissues often detach from a Millicell® membrane and can essentially float and move in the culture medium. As a consequence, imaging and tracking of individual cells within the tissue slice is made much more difficult leading to inaccurate measurements and is sometimes altogether impossible.
Intact tissue slices can also be maintained and kept alive on Alvetex Strata membranes for several weeks. The presentation of the membrane in the well insert provides an ideal platform for supporting tissue viability. In the absence of a vascular supply, the tissue is dependent on efficient exchange of gases, nutrients and waste products with the incubating culture medium. Again, the high levels of porosity exhibited by the Alvetex Strata mem-brane automatically promotes enhanced exchange by passive diffusion within a static culture environment.
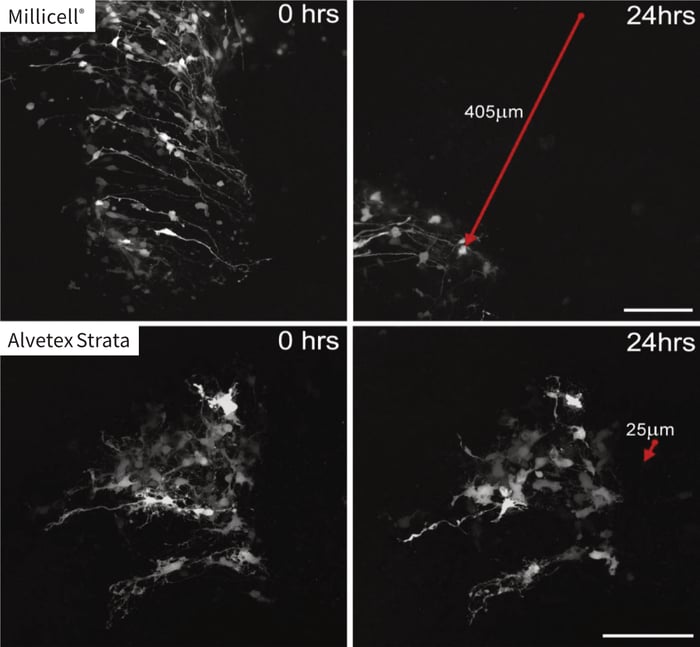
Figure 3. Time lapse imaging of spinal cord tissue slice and demonstration of tissue slippage. Images show the same field of view captured at time zero (A,C) and after 24 hours recording (B,D) for tissue slices maintained on either the Millicell® (A,B) or Alvetex Strata (C,D) porous membranes. Note how the cellular information captured in the field of view from the sample maintained on the Millicell® membrane has all but slipped out of frame during the 24 hour period. In contrast, samples main-tained on Alvetex Strata remained almost completely static. The red arrows show the direction and extent of tissue slippage observed. Scale bar: 100 microns. (Images courtesy of Kieran McDermott, University of Cork.)
Example Application 2: Maintenance of Stem-Cell Derived Embryoid Bodies on Alvetex Strata
Background
Pluripotent embryonic stem cells are recognized for their ability to self-renew and differentiate into cells constituting all three primitive germ layers (endoderm, mesoderm, ectoderm) and germ cells. Functional tests of pluripotency include: introduction of cells into the blastocyst resulting in a chimeric animal with embryonic stem cells contributing to all tissue types; formation of a teratoma xenograft comprising a mixture of differentiated tissues representative of all three germ layers; development of cell aggregates in suspension culture termed embryoid bodies. The latter in vitro approach of producing embryoid bodies avoids animal usage and can offer a more controlled for method certain experimental requirements (Bratt-Leal et al. 2009).
The culture of embryoid bodies derived from pluripotent stem cells provides the opportunity to study the early stages of embryogenesis in vitro in which certain aspects of three-dimensional tissue structures are produced. Signaling between cells is fundamental to normal dev-elopmental processes and is likely to be significantly constrained in conventional monolayer cultures. This is thought to be one of the primary reasons why differ-entiation by stem cells in vitro on flat two-dimensional substrates is very limited and does not reflect the extent of tissue differentiation observed in teratomas or embryoid bodies. Accordingly, the study of stem cell differentiation in three dimensional models has the potential to further our understanding of the basic mechanisms controlling the early stages of embryonic tissue development.
The growth of embryonic stem cells as embryoid bodies is well established and there are various methods of producing a suspended aggregate of stem cells, including hanging drops, static suspension cultures, entrapment of cells within a hydrogel, micro-fabricated substrates, and specialized devices. A review has been published describing several of the most common culture methods to produce embryoid bodies (Kurosawa, 2007). The application of the most basic approaches, namely hanging drops and static suspension cultures, were originally adopted for the assessment of in vitro differentiation of embryonal carcinoma stem cells (Martin and Evans, 1975). More recently, these and other technologies have been applied to investigate the developmental potential of human embryonic stem cells (Doetschman et al. 1985).
There are several ways of working with embryoid bodies, however, these are not without technical issues concerning the longer-term growth, maintenance and subsequent analysis. For example, exchanging or manipulating the small volume of medium (typically 20-30 μl) used for hanging drops is not without difficulty and the com-position of the medium cannot be easily controlled or assayed. This is especially relevant for longer-term culture and extended differentiation growth periods since it is important to maintain medium quality, and this is difficult in a small liquid volume. Working with static suspension cultures readily produces embryoid bodies but they frequently agglomerate forming large irregularly shaped masses of cells. Analysis of embryoid bodies is also challenging when working with a small piece of cellular material that is sometimes difficult to easily locate and handle.
Here we describe how Alvetex Strata can be used in combination with methods to produce embryoid bodies to address some of these technical issues and provide new opportunities for studying the differentiation of embryonic stem cells in vitro. We show how embryoid bodies can be maintained for long periods in vitro and their attachment to a porous substrate supports cell viability and improves ease of handling for subsequent downstream analysis.
Methodology
This is an example of using Alvetex Strata to support the growth and differentiation of embryoid bodies derived from human embryonic stem cells. This method outlines the nature and source of the materials and cells employed, and basic techniques used, as an assessment of the developmental potential of human embryonic stem cells in vitro.
- Embryoid bodies were derived from RC10 human embryonic stem cells provided by Roslin Cellab.
- RC10 cells were maintained as a feeder-free culture using CELLStart coating (Invitrogen) and StemPro human embryonic stem cell media kit (Invitrogen) in 6-well plates. The StemPro kit is based on DMEM/F12 with Glutamax supplemented with 25 % bovine serum albumin (BSA), StemPro growth supplement (50×) and 10 μg/ml basic fibroblast growth factor (bFGF). Complete media consists of 454ml DMEM/F12, 10 ml of 50× StemPro supplement, 36ml of 25% BSA to a give a final concentration of 8 % and finally 400 μl of the 10 μg/ml stock solution of bFGF to give a final concentration of 8 ng/ml.
- Cells were passaged using a manual dissociation technique and the resulting cell suspension was split between ×8 CELLStart coated wells. Media was changed every 48 hours until cultures reached 70-80 % confluency. Cells were split 1:8 once a week.
- Embryoid bodies were produced using the 800 micron Aggrewell plates supplied by Stem Cell Technologies as described by Antonchuk (2013). The media used for the formation and maintenance of embryoid bodies was Knock-Out DMEM (Invitrogen) supplemented with 50 ml human embryonic stem cell qualified FBS, 5 ml of 200 mM L-glutamine (Invitrogen); 5 ml Non-Essential Amino Acids (Invitrogen); and 1 ml 2-mercaptoethanol (Invitrogen) (EB media).
- The AggreWell™ plates contain 800 micron inverse pyramidal micro-wells – the size of the micro-wells determines the size of the embryoid bodies which are formed. Each well was washed with 1ml of PBS containing calcium and magnesium. This was replaced with 0.5 ml EB media and the plate was centrifuged at 2000g for 5 min to force air bubbles out of the wells. The plate was then placed in an incubator at 37 °C and 5 % CO2 to equilibrate while cells are harvested.
- A 70-80 % confluent RC-10 cell culture was selected for the formation of embryoid bodies. Prior to cell harvesting the ROCK inhibitor – Y-27632 (Tocris Bioscience) was added to the cultures to a final concentration of 10μm for 1 hour. This helps to prevent cell death when the cells are dissociated into a single cell suspension. A single cell suspension of was achieved by treating the cells with TrypLE™ Select CTS™ solution (Invitrogen) and cells were counted using a haemocytometer.
- Cells were added directly to the pre-warmed media in the AggreWell™ plates. For the formation of 800 micron diameter embryoid bodies, 9.0 × 105 cells were added to an AggreWell™ 800 plate containing 300 micro-wells. This formed embryoid bodies containing approximately 3000 cells each. The EB media was topped up to a final volume of 2ml per plate and supplemented with 10 μM Y-27632. The cells were pipetted up and down to evenly distribute the cells and the plate was immediately centrifuged at 300 g for 2 minutes to force the cells into the micro-wells.
- The cells were cultured for 48 hours at 37 °C and 5 % CO2. After 48 hours incubation, embryoid bodies are clearly visible within each micro-well. Embryoid bodies were harvested by firmly pipetting the medium up and down three times using a blue pipette tip to dislodge the embryoid bodies from the micro-wells. The resulting suspension was passed through a 40 μm cell strainer to remove any remaining single cells. The strainer was then inverted and fresh EB media was used to back wash the aggregates into a 50 ml falcon tube. Embryoid bodies were counted and approximately 1000 embryoid bodies per well were added to non-tissue culture treated 6 well plates. The embryoid bodies were then maintained for a further 7 days.
- Alvetex Strata membranes were hydrated using 70 % ethanol by adding enough 70% ethanol to completely cover the membranes and then and washed twice in PBS prior to use. Well inserts were topped up with 7 ml of EB differentiation media and placed in the incubator to equilibriate until required.
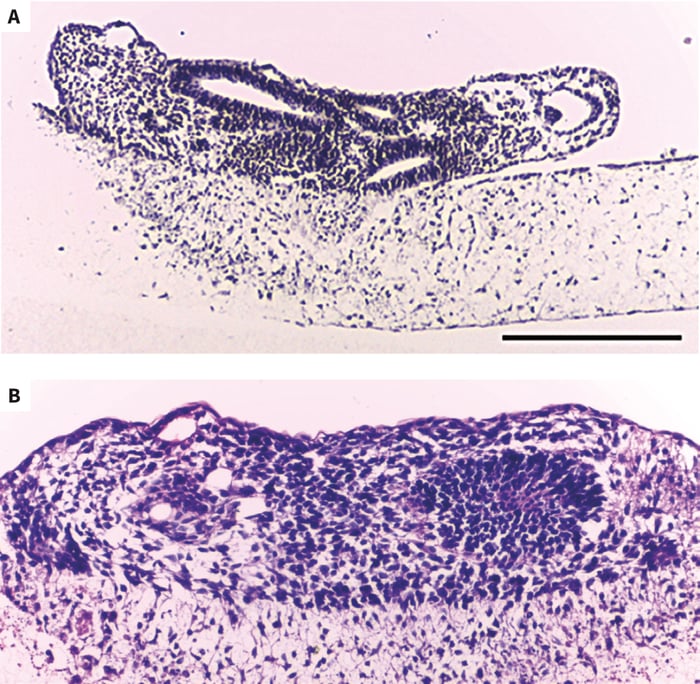
Figure 4. Light micrographs of human embryonic stem cell- derived embryoid bodies initially developed using an AggreWell plate and then subsequently maintained for 7 days on an Alvetex Strata membrane presented in a 6-well insert (A,B). Embryoid bodies were heterogeneous in structure and contained a diverse array of different cell types. Tissues were stained with Hematoxylin and Eosin. Scale bars: (A) 200μm; (B) 150μm.
- Embryoid bodies were collected from their suspension culture and allowed to settle in 15 ml centrifuge tubes, spent media was then removed, and then topped up with 1ml of fresh EB media. Embryoid bodies were distributed evenly over the surface of the Alvetex Strata membranes using a blue pipette tip.
- Embryoid bodies were cultured on Alvetex Strata membranes for a further 7 days un-disturbed in the same media before being fixed in 4 % paraform-aldehyde overnight. Care should be taken not to dislodge embryoid bodies if media changes are made.
- Fixed embryoid bodies on Alvetex Strata membranes were subsequently embedded in paraffin wax, sectioned on a microtome (10 μm) and mounted on microscope slides.
- To visualize the morphology of the embryoid bodies, tissue sections were stained with Hematoxylin and Eosin.
- To assess the presence of tissues representative of the three germ layers, immunocytochemistry was performed using antibodies reactive to markers of ectoderm, mesoderm and endoderm: namely, β-III- tubulin (TUJ1, 1:600, Cambridge Bioscience PRB-435P-100); smooth muscle actin (SMA, 1:100, Abcam ab5694); α-feto-protein (AFP, 1:500, Sigma Aldrich A8452), respectively.
NB. See Protocols for detailed protocols concerning tissue embedding, sectioning, staining and immunocytochemical analysis.
Results
These data demonstrate the suitability of using Alvetex Strata to support the long-term maintenance of embryoid bodies in culture. This approach is advantageous compared to maintaining embryoid bodies for long periods in suspension culture by the hanging drop technique for several practical reasons as follows:
Improved Cell Viability
The embryoid bodies slowly change shape and tend to flatten forming a disc-like structure up to 200μm thick over the surface of Alvetex Strata. Cells within these avascular structures are within a reasonable distance from the incubating medium to exchange nutrients and waste products by diffusion. It is sometimes an issue with spherical embryoid bodies that they become too large and necrotic centers can develop, leading to changes in cell differentiation and even cell death.
Alvetex Strata is a highly porous membrane which enables a good level of nutritional support for good embryoid body viability from beneath when using the well insert format. Conventional membranes presented in well inserts designed for feeding cells from beneath (e.g. Millicell®) are approximately three times less porous compared to Alvetex Strata.
Improved Cell Attachment
Secure attachment to the surface of the Alvetex Strata membrane also has its benefits when changing the cell culture medium. This is easily performed and minimizing loss, disturbance and damage to the embryoid body that can often happen when using hanging drop techniques. Moreover, hanging drops using either modern adapted culture plates or the conventional method, are limited by the volume of medium used. This is far less of an issue when working with an Alvetex Strata well insert where the volumes of medium used are greater and more easily controlled.
Versatility
Alvetex Strata is a versatile technology. Like conventional culture plastic, it can be coated with various standard reagents used in cell culture if required (e.g. collagen, fibronectin, Matrigel®, poly-d-lysine, etc.). Researchers can then investigate how embryoid bodies react to such substrates presented in different ways. Furthermore, an additional option includes the co-culture of different cell types on Alvetex Strata prior to seeding an embryoid body.
As demonstrated herein, tissue sections of the embryoid body are straightforward to produce. This enables assessment of basic tissue structures using histological methods but also more in-depth analysis of gene and protein expression using in situ hybridization and immunocytochemical methods, respectively. These techniques are useful for the assessment of cell differentiation and developmental potential of the stem cells used. The embryoid bodies produced in this example application were highly differentiated. They could have been cultured and maintained for several weeks longer to enable further differentiation. This may also be useful for researchers wanting to explore later stages of embryonic development.
Potential Replacement of Teratoma Assay
As described, there are numerous benefits for growing embryoid bodies on Alvetex Strata. It is also feasible that this approach could replace certain aspects of the conventional teratoma assay that is recognized as the gold standard to assessing the developmental potential of pluripotent stem cells. Many investigators use the teratoma approach to identify, most often by basic histological methods, tissues representative of all three germ layers. As demonstrated herein, this can be easily achieved in vitro using Alvetex Strata in combination with more accurate tissue markers. This method is not necessarily a complete replacement for the teratoma assay, but it does offer researchers an alternative, more rapid and controllable test, which could also result in reducing the number of animals used in research.
As the field of stem cell biology develops and the need to characterize the developmental potential of pluripotent stem cells increases, the growth, maintenance, and assessment of embryoid bodies on Alvetex Strata offers an alternative strategy to cell biologists working with embryonic stem cells and induced pluripotent stem cells (iPSCs).
Conclusions
Alvetex Strata is a highly porous polystyrene membrane presented in a well insert. The porous structure of Alvetex Strata enables efficient exchange of gases and solutes to support cell viability from beneath. The rough surface topography enhances the attachment of tissue pieces to the membrane, securing the sample and preventing slippage or tissue loss.
We present two different examples for the use of Alvetex Strata in the maintenance of intact tissue cyto-architecture in vitro. There are many other instances where this technology can be applied to extend the viability of organotypic tissue cultures and ease practical difficulties regarding sample handling.
References
- Antonchuk J (2013). Formation of embryoid bodies from human pluripotent stem cells using AggreWell plates. Methods in Molecular Biology (Clifton, N.J.) 946:523-533.
- Banker GA, Cowan WM (1977). Rat hippocampal neurons in dispersed cell culture. Brain Res 126:397-442.
- Bratt-Leal A, Carpenedo R, McDeitt TC (2009). Engineering the embryoid body microenvironment to direct embryonic stem cell differentiation. Biotechnolo Prog 25:43-51.
- Doetschman TC, Eistetter H, Katz M, Schmidt W, Kemler R (1985). The in vitro development of blastocyst- derived embryonic stem cell lines. J Embryol Exp Morphol 87:27- 45.
- Fisher RL, Vickers AE (2013). Preparation and culture of precision-cut organ slices from human and animal. Xenobiotica 43:8-14.
- Harrison RG (1907). Observation on the living developing nerve fiber. Anat Rec 1:116-18.
- Hogue MJ (1947). Human fetal brain cells in tissue culture: their identification and motility. J Exp Zool 1006:85-107.
- Galwhiler BH, Capogna M, Dehanne D, McKinney RA, Thompson SM (1997). Organotypic slice cultures: a technique has come of age. Trends Neurosci 20:471-7.
- Galwhiler BH, Thompson SM, Muller D (2001). Preparation and maintenance of organotypic slice cultures of CNS tissue. Curr Protoc Neurosci Chapt 6.
- Kurosawa H (2007). Methods for inducing embryoid body formation: in vitro differentiation system of embryonic stem cells. J Biosci Bioeng 103:389-98.
- Martin GR, Evans MJ (1975). Differentiation of clonal lines of teratocarcinoma cells: formation of embryoid bodies in vitro. Proc Natl Acad Sci USA 72:1441-45.
- Miyata T, Saito K, Nishizawa Y, Murayama A, Massaoka M, Ogawa M (2005). Modern slice culture for direct observation of production and migration of brain neurons. Nagoya J Med Sci 67:65-70.
- Nakai J (1956). Dissociated dorsal root ganglia in tissue culture. Am J Anat 99:81-130
- Stoppini L, Buchs PA, Muller D (1991). A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37:173-82.