Alvetex Scaffold Application Note 01
Formation of Mesenchymal Tissues in Alvetex Scaffold Derived From Stem Cells and Established Cell Lines
Download this application note as a PDF (3.49 MB)
Highlights:
- Sustained long-term culture of undifferentiated rat MSCs in Alvetex Scaffold
- Enhanced expression of osteogenic and adipogenic markers compared to 2D culture
- Formation of extracellular matrix proteins and bone nodules
- Compatibility with conventional analytical methods
Introduction
Based on an increasing body of evidence, it is generally accepted that culturing cells in three dimensional (3D) models can lead to superior cell viability, differentiation and function compared to existing conventional culture systems where cells largely grow as monolayers on two dimensional (2D) substrates. These enhanced qualities are important for the retention of the in vivo-like cell phenotype and cell differentiation potential and subsequent functionality as required for downstream applications. Here we demonstrate the application of Alvetex Scaffold technology for the growth and differentiation of mesenchymal cells and the formation of differentiated tissues.
Alvetex Scaffold is a novel substrate that enables a solution for simple and routine 3D culture. It is composed of a highly porous polystyrene scaffold that has been engineered into a 200 micron thick membrane to enable entry of cells and efficient exchange of gases and solutes. Cells enter the fabric of the scaffold, retain their natural 3D structure, and form close 3D interactions with adjacent cells. Unlike conventional 2D culture, cells in Alvetex Scaffold do not grow as monolayers and do not undergo the flattened shape transition that can result in aberrant changes to gene and protein expression and con-sequently cellular function.
Mesenchymal stem cells (MSCs) are adherent multipotent cells derived from tissue such as bone marrow and which possess the ability to differentiate in vitro into a number of tissue types including bone, cartilage and muscle [1]. In this application note we demonstrate that MSCs extracted from the bone marrow of adult rats can be successfully cultured in 3D in Alvetex Scaffold and induced to differentiate into osteogenic and adipogenic derivatives more efficiently than their 2D counterparts. We also report bone formation and the production of extracellular matrix by MG63 cells which represent an established cell line derived from a human osteosarcoma. The data generated here is supported by peer-reviewed literature [2,3] and clearly shows that Alvetex Scaffold promotes enhanced in vitro differentiation. Furthermore, the technology is convenient to use, as well as being compatible with a range of standard downstream analytical techniques commonly prac-tised in molecular and cellular biology.
3D Culture of Mesenchymal Stem Cells in Alvetex Scaffold
Populations of rat primary MSCs grown on Alvetex Scaffold showed a consistent and increasing growth pattern over the 14 day test period (Figure 1A). Cell viability was assessed by MTT assay. Absorbance values increased in a linear fashion and did not plateau during this period suggesting further growth potential. Staining the 3D culture with Neutral Red showed the gross distribution and density of MSCs in the scaffold (Figure 1B). After 14 days cells were located over the majority of the Alvetex Scaffold disc, although there remained some space for further expansion of the cell population around the periphery. Histological analysis revealed the microscopic distribution of 3D cells inside the scaffold (Figure 1C). MSCs show a relatively homogeneous distribution but do not pack closely together at this stage and therefore cell-cell contact inhibition of growth is avoided. This enables the continued 3D expansion of the cell population over the 14 day culture period.
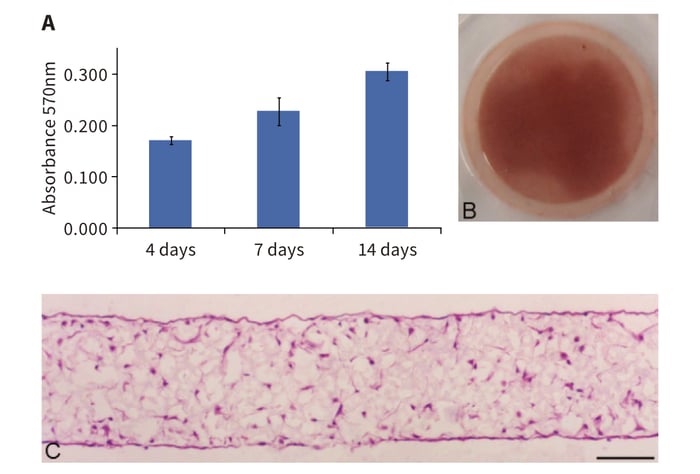 Figure 1: Alvetex Scaffold supports 3D culture of viable rat MSCs and the linear expansion of the cell population. (A) MTT viability assay of rat MSCs cultured in Alvetex Scaffold over 14 days. Data represent average absorbance at 570 nm, n = 3, ± SEM; (B) Gross view of Neutral Red staining of rat MSCs cultured in Alvetex Scaffold for 14 days; (C) Micrograph of H&E stained, wax-embedded sections from rat MSCs cultured in Alvetex Scaffold for 14 days. Scale bar: 100 μm.
Figure 1: Alvetex Scaffold supports 3D culture of viable rat MSCs and the linear expansion of the cell population. (A) MTT viability assay of rat MSCs cultured in Alvetex Scaffold over 14 days. Data represent average absorbance at 570 nm, n = 3, ± SEM; (B) Gross view of Neutral Red staining of rat MSCs cultured in Alvetex Scaffold for 14 days; (C) Micrograph of H&E stained, wax-embedded sections from rat MSCs cultured in Alvetex Scaffold for 14 days. Scale bar: 100 μm.
Osteogenic Differentiation of MSCs in Alvetex Scaffold
Cultured rat MSCs grown in 3D on Alvetex Scaffold were induced to differentiate into bone-forming cells following a standard procedure. Analysis of the expression of markers associated with osteogenesis was performed and demonstrated the differentiation of bone-producing cells (Figure 2). Compared to cells grown on 2D plastic, cells grown on Alvetex Scaffold deposited more calcium and maintained stable collagen production which is indicative of osteogenic rather than chondrogenic differentiation. Furthermore, the culture of MSCs on Alvetex Scaffold without any osteogenic stimulus was enough to induce the deposition of calcium (data not shown). The production of alkaline phosphatase in response to incubation in osteogenic medium was increased in cells growing on Alvetex Scaffold compared with cells cultured on 2D substrates. Alkaline phosphatase is involved in the regulation of phosphate availability for hydroxyapatite production [4]. It is likely therefore that the higher levels of alkaline phosphatase in 3D culture contributed to the greater amount of calcium deposited in Alvetex Scaffold cultures compared to their 2D counterparts. Osteocalcin was also greatly increased in cells differentiated on Alvetex Scaffold, which is associated with activation of insulin signalling and bone matrix remodelling via resorption in vivo [5].
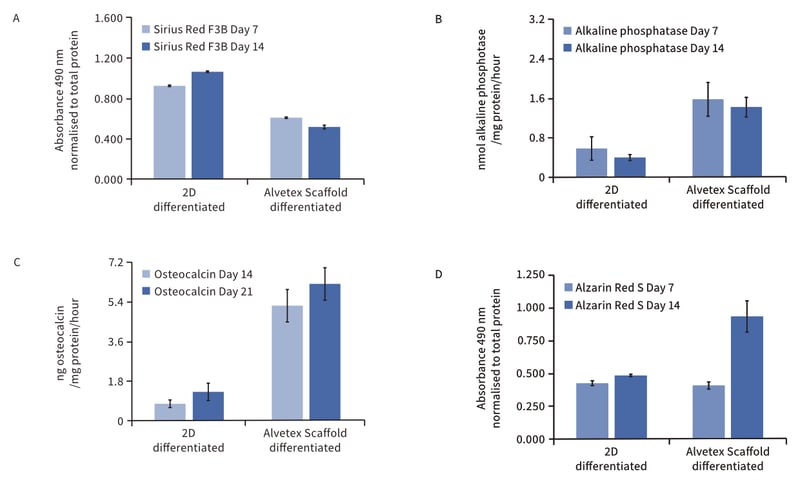 Figure 2: Cell growth on Alvetex Scaffold enhances osteogenic differentiation of rat MSCS compared to conventional 2D culture. Data plots show: (A) Sirius Red F3B staining for collagen. Bars represent ratio between average absorbance at 490 nm and average total protein content as determined by Bradford assay. n = 3, ± SEM; (B) Alkaline phosphatase activity as assayed by dephosphorylation of p-nitrophenyl phosphate substrate and normalised to average total protein content as determined by Bradford assay. Bars represent average nmol of alkaline phosphatase per mg of total protein per hour. n = 3, ± SEM; (C) ELISA detection for osteocalcin released into conditioned medium. Data normalised to average total protein content as determined by Bradford assay. Bars represent average ng of osteocalcin per mg of total protein per hour. n = 3, ± SEM; (D) Alizarin Red S staining for calcium. Bars represent ratio between average absorbance at 490nm and average total protein content as determined by Bradford assay, n = 3, ± SEM.
Figure 2: Cell growth on Alvetex Scaffold enhances osteogenic differentiation of rat MSCS compared to conventional 2D culture. Data plots show: (A) Sirius Red F3B staining for collagen. Bars represent ratio between average absorbance at 490 nm and average total protein content as determined by Bradford assay. n = 3, ± SEM; (B) Alkaline phosphatase activity as assayed by dephosphorylation of p-nitrophenyl phosphate substrate and normalised to average total protein content as determined by Bradford assay. Bars represent average nmol of alkaline phosphatase per mg of total protein per hour. n = 3, ± SEM; (C) ELISA detection for osteocalcin released into conditioned medium. Data normalised to average total protein content as determined by Bradford assay. Bars represent average ng of osteocalcin per mg of total protein per hour. n = 3, ± SEM; (D) Alizarin Red S staining for calcium. Bars represent ratio between average absorbance at 490nm and average total protein content as determined by Bradford assay, n = 3, ± SEM.
Histological analysis of rat MSCs differentiating into bone during 3D culture on Alvetex Scaffold was performed using standard tissue fixation, processing and staining procedures. Wax embedded sections were processed with different stains to show deposition of various materials associated with osteogenesis. Von Kossa staining showed the presence of calcium deposits whilst histological counterstaining demonstrated the presence of collagen (Figure 3). Histological detection of markers such as calcium and collagen supports the quantitative data presented in Figure 2, indicating bone formation in 3D culture using Alvetex Scaffold technology.
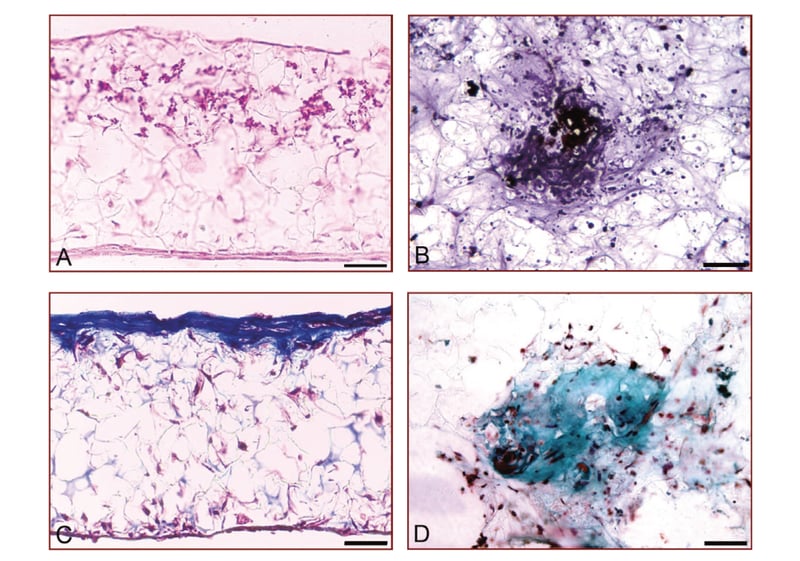 Figure 3: Deposition of calcium and collagen by rat MSCs differentiating into bone in 3D culture using Alvetex Scaffold. 3D cultures of rat MSCs grown on Alvetex Scaffold for 21 days. (A, B) Von Kossa silver staining to detect the deposition of calcium; (C, D) Masson’s Trichrome staining of extracellular collagen matrix. Note the deposition of significant amounts of collagen in the upper layers of the scaffold (C). Scale bars: 40 μm (A, C); 80 μm (B, D).
Figure 3: Deposition of calcium and collagen by rat MSCs differentiating into bone in 3D culture using Alvetex Scaffold. 3D cultures of rat MSCs grown on Alvetex Scaffold for 21 days. (A, B) Von Kossa silver staining to detect the deposition of calcium; (C, D) Masson’s Trichrome staining of extracellular collagen matrix. Note the deposition of significant amounts of collagen in the upper layers of the scaffold (C). Scale bars: 40 μm (A, C); 80 μm (B, D).
Differentiation of Human Osteosacroma Cells on Alvetex Scaffold
Alvetex Scaffold is a highly versatile technology able to support bone formation by different cell types including those derived from healthy or pathological bone tissues. For example, cell lines established from osteosarcomas are often used to study features of bone cell differentiation, function and disease. Here we also demonstrate the application of Alvetex Scaffold technology to support 3D culture of the osteosarcoma cell line, MG63 (Figure 4). These data demonstrate the ability of MG63 cells to grow and proliferate throughout the scaffold. Histological data (Figure 4A) shows that the pattern of MG63 cell growth closely resembles that seen with the MSCs (Figure 1C). Scanning electron microscopy reveals more clearly the complexity of MG63 cell morphology in 3D culture and the interactions between cells. Assessment of calcium deposition by Alizarin Red S staining was evident throughout the culture and easily detectable by standard light microscopy. Compared to MG63 cells grown on conventional 2D plastic, those grown on Alvetex Scaffold exhibited greater alkaline phosphatase activity and produced more osteocalcin.
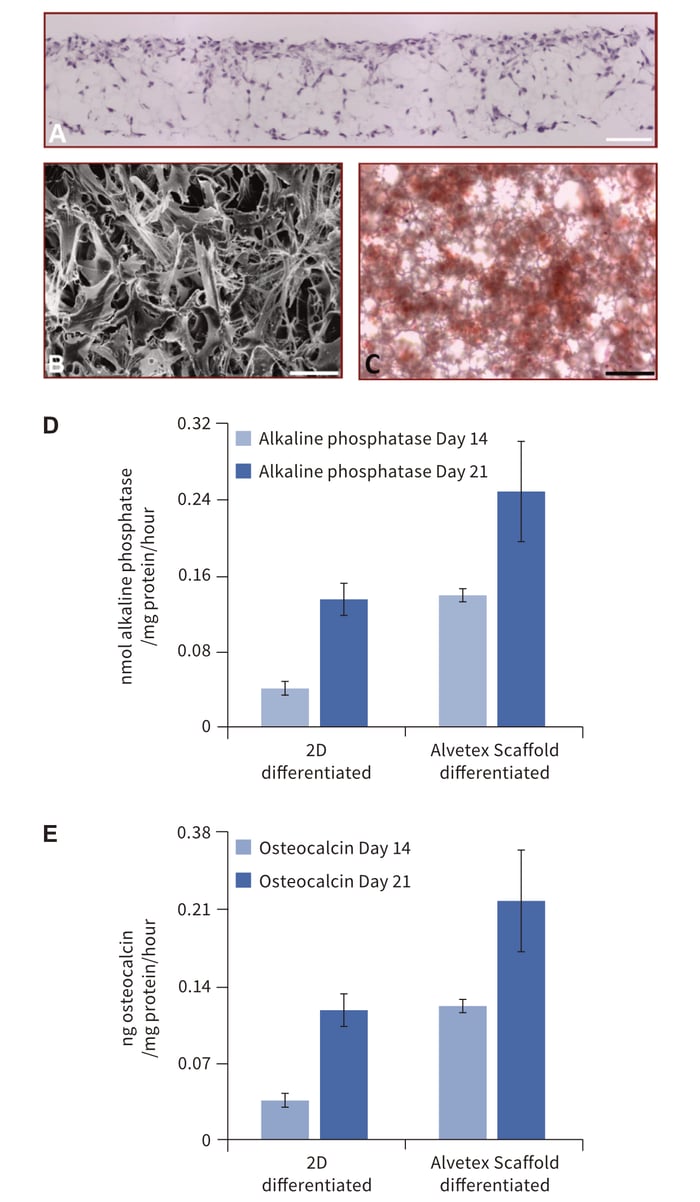 Figure 4. Growth and differentiation of MG63 cells on Alvetex Scaffold. (A) Micrograph of H&E stained, wax-embedded sections from MG63 cells cultured in Alvetex Scaffold for 7 days; (B) Scanning electron micrograph of MG63 cells cultured in Alvetex Scaffold as viewed from above; (C) Light micrograph of Alizarin Red S staining showing the gross distribution of calcium deposition by MG63 cells cultured in Alvetex Scaffold for 21 days. This image was taken from directly above the scaffold disc; (D) The expression of alkaline phosphatase activity in differentiating MG63 cells grown on Alvetex Scaffold for up to 21 days was assayed by dephosphorylation of p-nitrophenyl phosphate substrate, normalised to average total protein content. Data represent average nmol of alkaline phosphatase per mg of total protein per hour, n = 3, ± SEM; (E) The concentration of osteocalcin released into the medium was determined by ELISA and normalised to average total protein content. Data represent average ng of osteocalcin per mg of total protein, n = 3, ± SEM. Total protein levels were determined by Bradford assays. Scale bars: 100 μm (A); 20 μm (B); 50 μm (C).
Figure 4. Growth and differentiation of MG63 cells on Alvetex Scaffold. (A) Micrograph of H&E stained, wax-embedded sections from MG63 cells cultured in Alvetex Scaffold for 7 days; (B) Scanning electron micrograph of MG63 cells cultured in Alvetex Scaffold as viewed from above; (C) Light micrograph of Alizarin Red S staining showing the gross distribution of calcium deposition by MG63 cells cultured in Alvetex Scaffold for 21 days. This image was taken from directly above the scaffold disc; (D) The expression of alkaline phosphatase activity in differentiating MG63 cells grown on Alvetex Scaffold for up to 21 days was assayed by dephosphorylation of p-nitrophenyl phosphate substrate, normalised to average total protein content. Data represent average nmol of alkaline phosphatase per mg of total protein per hour, n = 3, ± SEM; (E) The concentration of osteocalcin released into the medium was determined by ELISA and normalised to average total protein content. Data represent average ng of osteocalcin per mg of total protein, n = 3, ± SEM. Total protein levels were determined by Bradford assays. Scale bars: 100 μm (A); 20 μm (B); 50 μm (C).
Adipogenic Differentiation of MSCs in Alvetex Scaffold
We have demonstrated the application of Alvetex Scaffold to support the differentiation of bone. The application of Alvetex Scaffold to support adipogenesis and the formation of fat-producing cells was also investigated (Figure 5). Rat MSCs were grown on Alvetex Scaffold and induced to differentiate toward the adipogenic lineage using a standard method. Samples of the differentiated cells were then successfully retrieved from the scaffold and subsequently cytospun onto microscope slides and stained for analysis. The data show a greater number of lipid droplets present in individual cells grown in 3D culture using Alvetex Scaffold compared to equivalent cells grown in conventional 2D culture. This observation was confirmed and quantified using an alternative approach that measured the absorbance of Oil Red O staining in samples extracted directly from the two culture methods. The data were normalised to take into account any differences in cell numbers. Overall, the results demonstrate that MSCs differentiating toward the adipogenic lineage produce greater quantities of lipid when grown in 3D culture compared to their counterparts maintained in conventional 2D plastic-ware. Such differences were detectable after 7 days after induction of differentiation and were maintained for up to 21 days. This outcome suggests that 3D culture of rat MSCs favours improved cell growth and differentiation.
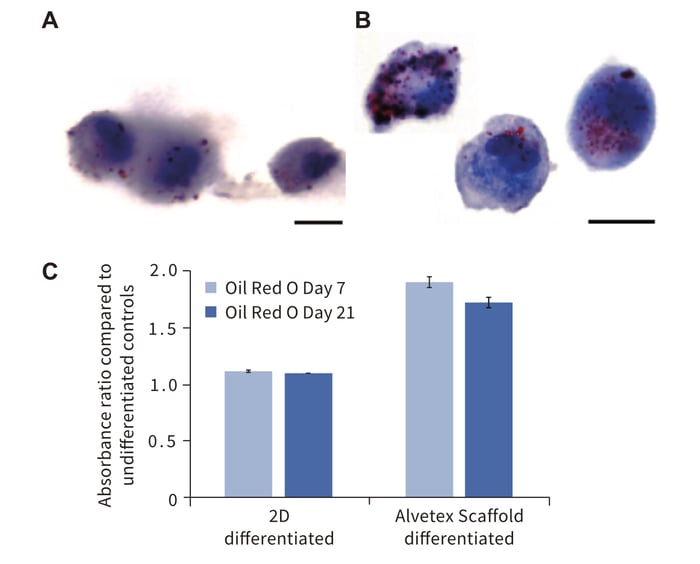 Figure 5: Cell growth on Alvetex Scaffold enhances adipo-genic differentiation of rat MSCS compared to conventional 2D culture. Data show: Oil Red O staining for lipid droplets in cells grown in (A) conventional 2D culture, or (B) 3D culture on Alvetex Scaffold. Cells were detached from their corresponding growth substrate using trypsin and sub-sequently cytospun onto glass slides prior to staining; (C) Quantitation of Oil Red O staining for lipids isolated from cells grown in 2D or 3D culture over 21 days. Data represent the average ratio of absorbances at 490nm between differentiated and undifferentiated cultures, n = 3, ± SEM. Scale bars: 10 μm (A,B).
Figure 5: Cell growth on Alvetex Scaffold enhances adipo-genic differentiation of rat MSCS compared to conventional 2D culture. Data show: Oil Red O staining for lipid droplets in cells grown in (A) conventional 2D culture, or (B) 3D culture on Alvetex Scaffold. Cells were detached from their corresponding growth substrate using trypsin and sub-sequently cytospun onto glass slides prior to staining; (C) Quantitation of Oil Red O staining for lipids isolated from cells grown in 2D or 3D culture over 21 days. Data represent the average ratio of absorbances at 490nm between differentiated and undifferentiated cultures, n = 3, ± SEM. Scale bars: 10 μm (A,B).
Conclusions
This application note describes the use of Alvetex Scaffold to support the differentiation of mesenchymal cells towards osteogenic and adipogenic phenotypes by providing a 3D cell culture environment. Both MSCs and MG63 cells readily grow and proliferate in the scaffold, establishing genuine 3D cultures. The data presented demonstrates that 3D culture using Alvetex Scaffold radically improves the ability of these cells to form mesenchymal tissues such as bone and fat. Superior cell morphology is achieved in 3D culture that is likely to contribute to the enhanced cell function as seen by increased levels of osteogenic and adipogenic markers. Cells from primary sources and established cell lines have been used which underlines the versatility of Alvetex Scaffold technology for alternative applications. A range of different analytical methods has been used to test and evaluate the status of cell growth and differentiation, highlighting the compatibility of Alvetex Scaffold with standard molecular and cellular techniques. Overall, Alvetex Scaffold provides a simple and robust approach for routine 3D cell culture. It is a convenient tool for promoting both osteogenic and adipogenic differentiation by mesenchymal cells. The highly porous structure of Alvetex Scaffold and range of alternative formats provide new opportunities for maintaining cells in 3D culture and creating more physio-logically relevant in vitro models and the formation of tissue-like structures.
References
- Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science, 1999. 284(5411): p 143-7.
- Neofytou EA, Chang E, Patlola B, et al. Adipose tissue-derived stem cells display a proangiogenic phenotype on 3D scaffolds. J Biomed Mat Res Part A, 2011. 98(3): p 383-93.
- Bokhari M, Carnachan RJ, Przyborski SA, and Cameron NR. Emulsion-templated porous polymers as scaffolds for three dimensional cell culture: effect of synthesis parameters on scaffold formation and homogeneity. J Mater Chem, 2007. 17: p 4088-94.
- Orimo H. The mechanism of mineralization and the role of alkaline phosphatase in health and disease. J Nippon Med Sch, 2010. 77(1): p 1-12.
- Ducy P. The role of osteocalcin in the endocrine crosstalk between bone remodelling and energy metabolism. Diabetologia, 2011. 54: p 1291-7.
- Croft AP, and Przyborski SA, Formation of neurons by non-neural adult stem cells: potential mechanism implicates an artefact of growth in culture. Stem Cells, 2006. 24(8): p 1841-51.
Methods
Detailed protocols for MTT viability assay, neutral red staining, H&E staining and SEM imaging are available in Alvetex Protocols.
Cell culture
Primary rat MSCs were derived from the bone marrow of 6-8 week old male Wistar rats (Harlan) using standard, previously published, methodology [6], and used at P1 for the experiment. Human osteosarcoma MG63 cells were obtained from ATCC (CRL-1427). Alvetex Scaffold 6-well inserts (product code AVP004) presented in 6-well plates were prepared for inoculation by ethanol wetting followed by two PBS rinses. The second PBS rinse was aspirated before cells were inoculated onto the polymer. A single cell suspension was prepared using trypsin following standard methods. Cells were counted on a haemocytometer, centrifuged and resuspended in a sufficient volume of medium to give a concentration of 10 million cells/ml. Cells were inoculated onto prepared Alvetex Scaffold discs as a 100 μl droplet of 1 million cells pipetted into the centre of the polymer disc. Inoculated discs were placed in a cell culture incubator (5 % CO2, 37°C) for 15 minutes to allow the cells to begin to attach to Alvetex Scaffold. Wells were then filled with 10 ml growth medium (DMEM High glucose supplemented with 10 % heat inactivated FCS; 1× non-essential amino acids; 2 μM L-glutamine; 100 U/ml penicillin and 100 U/ml streptomycin). Undifferentiated 3D cultures were maintained in the growth medium. For osteogenic differentiation, cells were incubated for approximately 4 days in growth medium, after which cultures were switched to osteogenic medium that comprised of growth medium supplemented with 100 nM dexamethasone, 50 μM ascorbic acid-2-phosphate and 10 nM β-glycerophosphate. The medium was changed for all cultures every 3-4 days. For adipogenic differentiation cells were incubated for approximately 4 days in growth medium, after which cultures were induced to differentiate by supplementing the growth medium in an induction/maintenance cycle. Induction medium was normal DMEM as detailed above, supplemented with 1 μM dexamethasone, 0.2 mM indomethacin, 10 μg/ml insulin and 0.5 mM 3-isobutyl-1-methylxanthine. Maintenance medium was normal DMEM as detailed above, supplemented with 10 μg/ml insulin. The induction/maintenance cycle ran as 3 days induction / 2 days maintenance for a total of 3 weeks.
Markers of Osteogenesis
Calcium
Alizarin Red S is an anthraquinone derivate that binds calcium through chelation and is therefore used to detect calcium deposits in osteogenic cultures. Cell cultures were fixed in 4 % paraformaldehyde, washed twice in PBS and incubated with 1 ml of Alizarin Red S stain (2 % w/v Alizarin Red S in water pH 4.1-4.3), shaking at 50 rpm for 30 minutes at room temperature. Cultures were then washed with PBS 3 times for 10 minutes with shaking as above. Cultures were then de-stained for quantitation by adding 1 ml 5 % perchloric acid, shaking at 50 rpm for 10 minutes and reading 200 μl samples in a plate reader at 490 nm. Silver is a versatile staining agent and is used as part of Von Kossa staining to detect calcium phosphate deposits in bone. Cell cultures were fixed in 4 % paraformaldehyde, embedded in paraffin and sectioned according to protocols available at Alvetex Protocols. Sections were deparaffinised and hydrated to water, prior to incubation in 1 % silver nitrate solution under UV for 1 hour. After rinsing in water, unreacted silver was removed by washing with 5 % sodium thiosulfate for 5 minutes, followed by further water rinses and counterstaining with fast red for 5 minutes. After a final rinse in water, sections were dehydrated, cleared and mounted for imaging.
Collagen
Sirius Red F3B (also called Direct Red 80) is a polyazo dye that recognises all types of collagen and exhibits yellow-to-green birefringence of collagen fibres. Cell cultures were fixed in 4 % paraformaldehyde, washed twice in PBS and incubated overnight with 1 ml of 1 mg/ml Direct Red 80 in saturated aqueous picric acid. Cultures were washed with PBS twice for 10 minutes at 50 rpm to remove excess stain, de-stained for quantitation using 1 ml 1:1, 0.2 M sodium hydroxide: methanol for 30 minutes with agitation and 200 μl samples were read in a plate reader at 490 nm.
Masson’s Trichrome staining is a well-characterised technique allowing the sequential staining of nuclei, cytoplasm and collagen fibres within a tissue section. Cell cultures were fixed in Bouin’s fixative, embedded in paraffin and sectioned according to protocols available at Alvetex Protocols. Sections were deparaffinised and hydrated to water prior to being incubated in Weigerts Iron Haematoxylin for 20-30 minutes, followed by careful rinsing in slow-running tap water for 10 minutes and incubated in Biebrich’s Scarlet for 10 minutes. Sections were again rinsed in water before being incubated in phosphomolybdic/phosphotungstic acid for 15 minutes, and immediately transferred to aniline blue for 7 minutes. After rinsing in water, sections were dipped into 1% acetic acid for 3 minutes, followed by a further water rinse. Finally, sections were quickly dehydrated, then cleared and mounted for imaging.
Osteocalcin
Osteocalcin is a small, insulin-regulating, non-collagenous protein secreted by osteoblasts and stored in bone through its binding to hydroxyapatite. Samples of conditioned medium were collected at appropriate time points and assessed for osteocalcin production by ELISA (Rat Osteocalcin Elisa Kit, BT-490, Biomedical Technologies Inc.) following manufacturer’s instructions and read in a plate reader at 450 nm.
Alkaline phosphatase
Alkaline phosphatase (ALP) hydrolyses inorganic pyro-phosphate (PPi) into inorganic phosphate (Pi), thus promoting hydroxyapatite formation. Quantification of ALP activity was achieved by utilising the de-phosphorylation of the phos-phatase substrate p-nitrophenyl phosphate. Cell cultures were lysed using lysis buffer (1 % v/v Igepal® CA-630; 10 mM Tris-HCl; 1 mM MgCl2, pH 7.5) with the assistance of an overnight freezing step at –80°C. Lysates were then cleared by centrifugation at 12,000 rpm for 10 minutes, after which 50 μl of supernatant were added to 50 μl p-nitrophenyl phosphate solution, wrapped in foil to exclude light, and incubated with shaking at 100 rpm for 30 minutes. Samples were read in a nanodrop spectrophotometer at 405 nm.
Normalisation
Total protein quantification was used to normalise data. Cell cultures were lysed using lysis buffer, freezing and clearing by centrifugation as described above. 10 μl of supernatant were then assayed using the Bradford assay to determine total protein yield.
Markers of Adipogenesis
Lipid droplets
Oil Red O is a fat-soluble diazo dye commonly used to detect lipids and lipid-filled vacuoles. To quantify lipid production, cell cultures were rinsed twice with PBS and fixed in 4 % para-formaldehyde for 10 minutes to 1 hour. 200 μl Oil Red O at 0.3 % w/v in 60 % isopropanol was added to each scaffold and incubated for 30 minutes at room temperature. Excess stain was removed by washing twice with 60 % isopropanol, then five times with distilled water. For quantitation, the cultures were de-stained in 1 ml 60 % isopropanol for 30 minutes with agitation at 50 rpm. 200 μl samples were read in a plate reader at 490 nm. To visualise lipid droplets, cell cultures were washed in PBS to remove the growth medium and cut into several pieces before placing in a 5 ml vial with 1 ml trypsin. The vials were put on a rotating disc in the cell culture incubator for 10 minutes, the trypsin solution containing the cut pieces was then removed into a small volume (approximately 4 ml) of fresh medium to neutralise the trypsin. This procedure was repeated twice, collecting the cells into a single tube for each polymer disc. Cells were then pelleted and re-suspended in 200 μl fresh medium. The cells were then cytospun onto glass slides following the manufacturer’s instructions at 800 rpm for 5 minutes and fixed overnight in 4 % paraformaldehyde before incubating in 0.3 % w/v Oil Red O in isopropanol for 30 minutes. After rinsing thoroughly with distilled water, cells were immediately visualised by light microscopy.